2 Control de calidad
Dra. Evelia Coss
07 de agosto de 2023
2.1 Material y Diapositivas
Diapositivas de Peter Hickey: Ve las diapositivas aquí.
Curso 2021 impartido por Leonardo Collado Torres: Ver información aquí
2.2 Enfoques o Ideas principales
Detectar células verdaderas y de alta calidad ✅🍪, de modo que cuando agrupemos nuestras células sea más fácil identificar poblaciones de tipos celulares distintos.
Identificar las muestras fallidas e intentar salvar los datos o eliminarlas del análisis, además de intentar comprender por qué falló la muestra.
2.3 ¿Por qué hay problemas con los datos?
- Problemas con el rompimiento de las células.
- Fallos en la preparación de las bibliotecas (ineficiencias en la transcripción inversa, amplificacion por PCR, etc).
- Más de una célula durante la secuenciación (douplets o multiplets).
- Problemas en el alineamiento.
2.4 Preguntas Básicas en Control de Calidad
Se responden durante el análisis de SingleCell RNA-Seq.
- ¿Cuántos droplets traen más de una célula? (douplets 🍪🍪 o multiplets 🍪🍪🍪).
- ¿Cuántas células murieron durante el proceso de secuenciacion?
Cada droplets debe contener una sola célula 🍪.
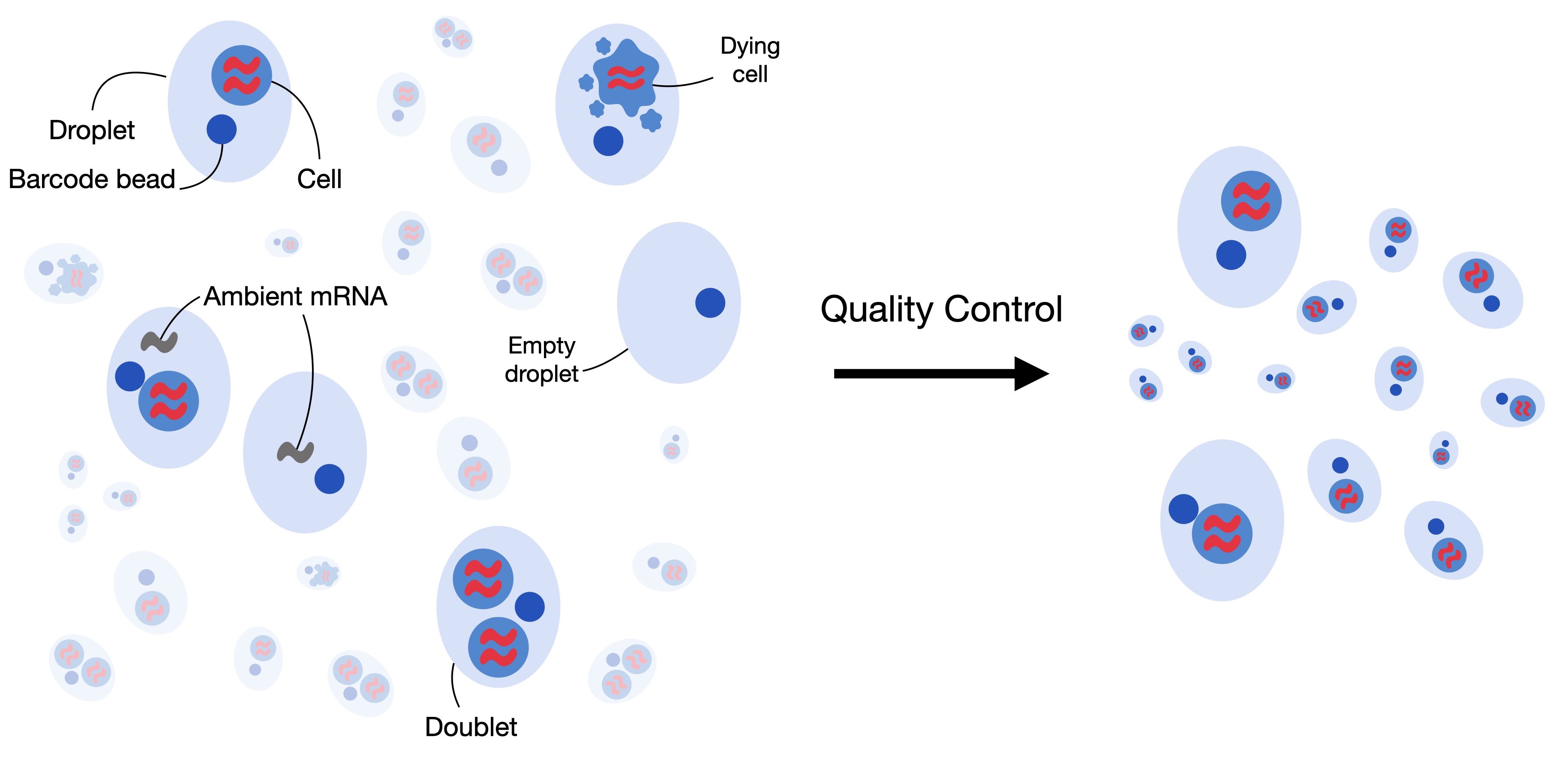 Figura obtenida de Single-Cell best practices.
Figura obtenida de Single-Cell best practices.
2.5 La mala calidad en los datos puede ser debida a varios factores
Una o varias células podemos encontrar:
- ⚠️ Baja cantidad de cuentas totales
- ⚠️ Baja cantidad de genes expresados
- ⚠️ Alta proporción de cuentas (reads) provenientes de la mitocondria
- ⚠️ Alta proporción de cuentas provenientes de las secuencias control (ERCC spike-in control transcripts)
Mas informacion sobre ERCC spike-in control transcripts.
2.6 Recomendaciones
- Tener un correcto diseño experimental (evitar efecto bash). 👾
- Correcta preparación de las muestras. 🎮
- Analizar la calidad de los datos. 🍪
2.7 Tipos de filtros
- Fixed thresholds: Valores de corte fijos y estrictos (FDR < 0.5) ♠️
- Adaptative thresholds: Valores de cortes adaptados al comportamiento de nuestros datos.♦️
2.8 Parámetros empleados para evaluar la calidad con la función addPerCellQC
La función addPerCellQC provenie del paquete scater. Agrega las siguientes métricas en cada célula y por cada gen dentro del mismo archivo.
sum: Número de cuentas (lecturas) totales de cada célula.detected: Genes expresados con al menos una cuenta.altexps_ERCC_percent: Porcentaje de cuentas mapeadas de las secuencias control (ERCC spike-in control transcripts).subsets_Mito_percent: Porcentaje de cuentas mapeadas provenientes de la mitocondria.
2.9 Ejemplo: linea celular 416 en ratón
Realizaremos las siguientes actividades con este ejemplo:
- Eliminar células de baja calidad.
- Comparar entre los tipos de filtros (Fixed thresholds y Adaptative thresholds).
- Filtrar células baja calidad.
- Visualización de datos crudos y filtrados.
 Línea celular de células mieloides progenitoras inmortalizadas de ratón usando SmartSeq2.
Línea celular de células mieloides progenitoras inmortalizadas de ratón usando SmartSeq2.
2.9.1 Paquetes e Importar los datos en R
Los paquetes que vamos a emplear para esta sección son:
library(scRNAseq) ## para descargar datos de ejemplo
library(DropletUtils) ## para detectar droplets
library(Matrix) ## para leer datos en formatos comprimidos
library(AnnotationHub) ## para obtener información de genes
library(scater) ## para gráficas y control de calidad
library(BiocFileCache) ## para descargar datos
library(EnsDb.Hsapiens.v86) ## Archivo de anotacion en humanos en Ensembl
library(dplyr) ## Modificacion de archivos dataframeCargar los datos empleando el paquete scRNAseq
## snapshotDate(): 2023-04-24## see ?scRNAseq and browseVignettes('scRNAseq') for documentation## loading from cache## see ?scRNAseq and browseVignettes('scRNAseq') for documentation## loading from cache## see ?scRNAseq and browseVignettes('scRNAseq') for documentation## loading from cache## snapshotDate(): 2023-04-24## see ?scRNAseq and browseVignettes('scRNAseq') for documentation## loading from cache## snapshotDate(): 2023-04-24## loading from cacheEl paquete scRNAseq contiene múltiples data sets compilados en funciones, para saber más da click aquí.
2.9.2 Anotación de genes
## snapshotDate(): 2023-04-24## AnnotationHub with 1 record
## # snapshotDate(): 2023-04-24
## # names(): AH73905
## # $dataprovider: Ensembl
## # $species: Mus musculus
## # $rdataclass: EnsDb
## # $rdatadateadded: 2019-05-02
## # $title: Ensembl 97 EnsDb for Mus musculus
## # $description: Gene and protein annotations for Mus musculus based on Ensembl version 97.
## # $taxonomyid: 10090
## # $genome: GRCm38
## # $sourcetype: ensembl
## # $sourceurl: http://www.ensembl.org
## # $sourcesize: NA
## # $tags: c("97", "AHEnsDbs", "Annotation", "EnsDb", "Ensembl", "Gene", "Protein", "Transcript")
## # retrieve record with 'object[["AH73905"]]'# Anotacion de genes con la localizacion de cada cromosoma
ens.mm.v97 <- ah[["AH73905"]] # solo un cromosoma## loading from cache## Warning: Unable to map 563 of 46604 requested IDs.2.9.3 Análisis de calidad con addPerCellQC
Al imprimir la variable sce.416b podemos observar que es de tipo SingleCellExperiment.
# Agregar la informacion de la calidad por cada gen y celula en un mismo archivo
# Identificar genes mitocondriales
sce.416b <- addPerCellQC(sce.416b,
subsets = list(Mito = is.mito)
)
sce.416b## class: SingleCellExperiment
## dim: 46604 192
## metadata(0):
## assays(1): counts
## rownames(46604): ENSMUSG00000102693 ENSMUSG00000064842 ... ENSMUSG00000095742 CBFB-MYH11-mcherry
## rowData names(1): Length
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(21): Source Name cell line ... altexps_SIRV_percent total
## reducedDimNames(0):
## mainExpName: endogenous
## altExpNames(2): ERCC SIRVPodemos observar la informacion de este data frame usando colData:
## DataFrame with 192 rows and 21 columns
## Source Name cell line cell type single cell well quality
## <character> <character> <character> <character>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 SLX-9555.N701_S502.C.. 416B embryonic stem cell OK
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 SLX-9555.N701_S503.C.. 416B embryonic stem cell OK
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 SLX-9555.N701_S504.C.. 416B embryonic stem cell OK
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 SLX-9555.N701_S505.C.. 416B embryonic stem cell OK
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 SLX-9555.N701_S506.C.. 416B embryonic stem cell OK
## ... ... ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S505... 416B embryonic stem cell OK
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S506... 416B embryonic stem cell OK
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S507... 416B embryonic stem cell OK
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S508... 416B embryonic stem cell OK
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S517... 416B embryonic stem cell OK
## genotype phenotype strain spike-in addition
## <character> <character> <character> <character>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype B6D2F1-J ERCC+SIRV
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype B6D2F1-J ERCC+SIRV
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. wild type phenotype B6D2F1-J ERCC+SIRV
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. induced CBFB-MYH11 o.. B6D2F1-J ERCC+SIRV
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 Doxycycline-inducibl.. induced CBFB-MYH11 o.. B6D2F1-J ERCC+SIRV
## ... ... ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl.. induced CBFB-MYH11 o.. B6D2F1-J Premixed
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl.. induced CBFB-MYH11 o.. B6D2F1-J Premixed
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl.. induced CBFB-MYH11 o.. B6D2F1-J Premixed
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl.. induced CBFB-MYH11 o.. B6D2F1-J Premixed
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl.. wild type phenotype B6D2F1-J Premixed
## block sum detected subsets_Mito_sum subsets_Mito_detected
## <factor> <numeric> <numeric> <numeric> <numeric>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 20160113 865936 7618 78790 20
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 20160113 1076277 7521 98613 20
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 20160113 1180138 8306 100341 19
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 20160113 1342593 8143 104882 20
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 20160113 1668311 7154 129559 22
## ... ... ... ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 20160325 776622 8174 48126 20
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 20160325 1299950 8956 112225 25
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 20160325 1800696 9530 135693 23
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 20160325 46731 6649 3505 16
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 20160325 1866692 10964 150375 29
## subsets_Mito_percent altexps_ERCC_sum altexps_ERCC_detected altexps_ERCC_percent
## <numeric> <numeric> <numeric> <numeric>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 9.09882 65278 39 6.80658
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 9.16242 74748 40 6.28030
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 8.50248 60878 42 4.78949
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 7.81190 60073 42 4.18567
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 7.76588 136810 44 7.28887
## ... ... ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 6.19684 61575 39 7.17620
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 8.63302 94982 41 6.65764
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 7.53559 113707 40 5.81467
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 7.50037 7580 44 13.48898
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 8.05569 48664 39 2.51930
## altexps_SIRV_sum altexps_SIRV_detected altexps_SIRV_percent total
## <numeric> <numeric> <numeric> <numeric>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 27828 7 2.90165 959042
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 39173 7 3.29130 1190198
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 30058 7 2.36477 1271074
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 32542 7 2.26741 1435208
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 71850 7 3.82798 1876971
## ... ... ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 19848 7 2.313165 858045
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 31729 7 2.224004 1426661
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 41116 7 2.102562 1955519
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 1883 7 3.350892 56194
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 16289 7 0.843271 1931645## [1] "Source Name" "cell line" "cell type" "single cell well quality"
## [5] "genotype" "phenotype" "strain" "spike-in addition"
## [9] "block" "sum" "detected" "subsets_Mito_sum"
## [13] "subsets_Mito_detected" "subsets_Mito_percent" "altexps_ERCC_sum" "altexps_ERCC_detected"
## [17] "altexps_ERCC_percent" "altexps_SIRV_sum" "altexps_SIRV_detected" "altexps_SIRV_percent"
## [21] "total"2.9.4 Preguntas sobre los datos
- ¿Cuántas células detectadas se encuentran en el dataframe
sce.416b? 1 - ¿Cuántos genes fueron analizados con al menos una cuenta? 2
Podemos conocer estas respuestas observando la informacion contenida en sce.416b, en dimensiones.
2.9.5 Visualización de los datos crudos
Genes detectados en cada uno de los bloques de secuenciación.
Genes detectados por cada tratamiento en cada bloque de secuenciación.
plotColData(sce.416b,
x = "block",
y = "detected",
other_fields = "phenotype"
) +
scale_y_log10() + # Cambiar la escala en Y a logaritmo
facet_wrap(~phenotype) + # Dividir tratamiento
labs(x = "Bloques de secuenciación", y="Genes detectados \n(log)", title = "Genes detectados") # Etiquetas en X, Y y titulo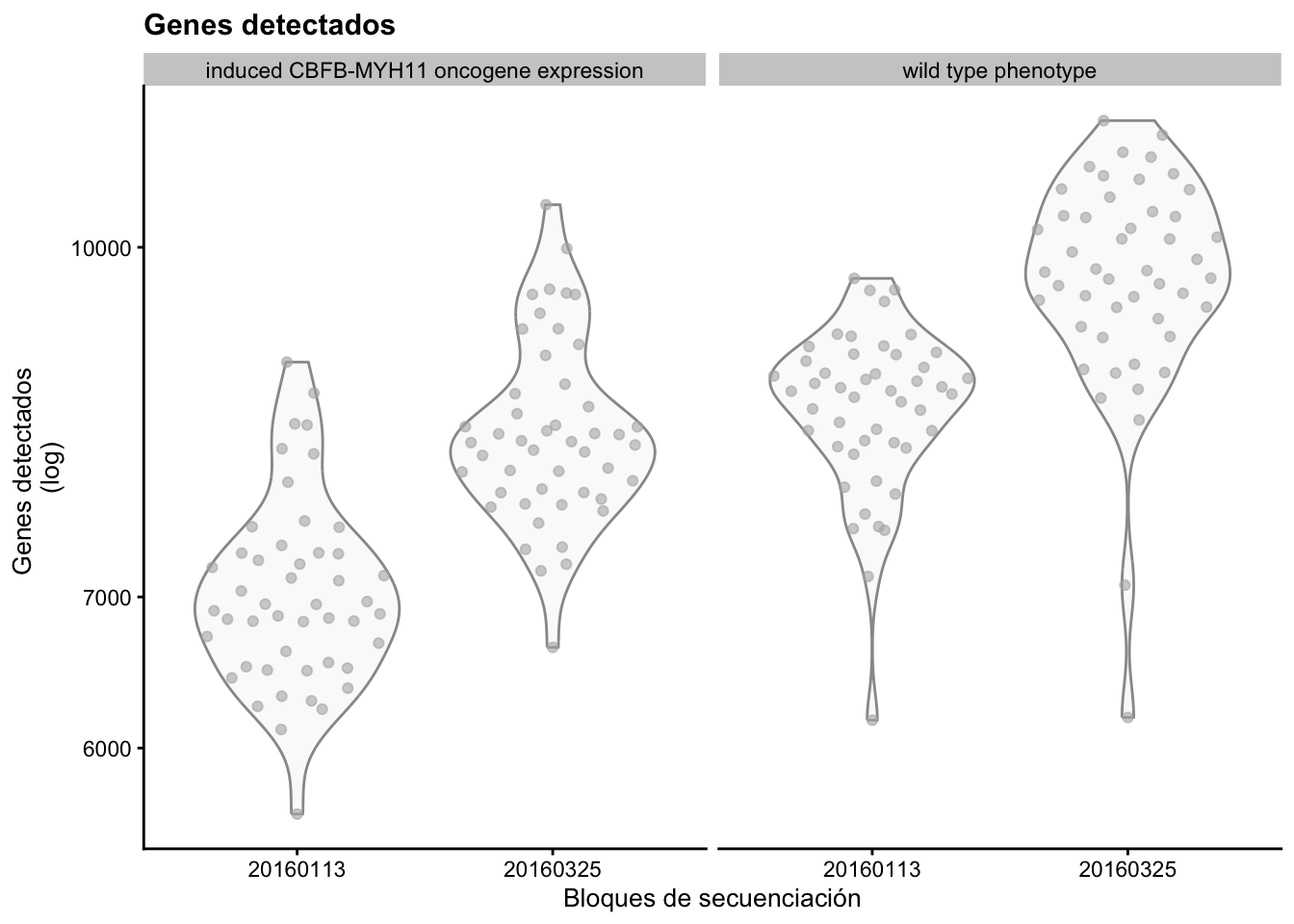
2.9.6 Filtro A: Fixed thresholds
Para el filtrado y eliminación de las células de baja calidad emplearemos las variables sum, detected, altexps_ERCC_percent y
subsets_Mito_percent.
# --- Fixed thresholds ---
# Valores de corte fijos y estrictos
qc.lib <- sce.416b$sum < 100000 # menos de 100 mil cuentas (lecturas)
qc.nexprs <- sce.416b$detected < 5000 # menos de 5 mil genes
qc.spike <- sce.416b$altexps_ERCC_percent > 10 # 10 % de las cuentas alineando a ERCC
qc.mito <- sce.416b$subsets_Mito_percent > 10 # 10 % alineando al genoma mitocondrial
discard <- qc.lib | qc.nexprs | qc.spike | qc.mito # si falla alguno de estos eliminarlo
# secuencias control = (ERCC spike-in control transcripts)
# Resumen del número de células
# Número de células eliminadas por cada filtro
DataFrame(
LibSize = sum(qc.lib),
NExprs = sum(qc.nexprs),
SpikeProp = sum(qc.spike),
MitoProp = sum(qc.mito),
Total = sum(discard)
)## DataFrame with 1 row and 5 columns
## LibSize NExprs SpikeProp MitoProp Total
## <integer> <integer> <integer> <integer> <integer>
## 1 3 0 19 14 332.9.7 Filtro B: Adaptative thresholds
Valores de cortes adaptados al comportamiento de nuestros datos.
# --- Adaptative thresholds ---
## Usando isOutlier() para determinar los valores de corte
qc.lib2 <- isOutlier(sce.416b$sum, log = TRUE, type = "lower")
qc.nexprs2 <- isOutlier(sce.416b$detected,
log = TRUE,
type = "lower"
)
qc.spike2 <- isOutlier(sce.416b$altexps_ERCC_percent,
type = "higher"
)
qc.mito2 <- isOutlier(sce.416b$subsets_Mito_percent,
type = "higher"
)
discard2 <- qc.lib2 | qc.nexprs2 | qc.spike2 | qc.mito2
# Extraemos los límites de valores (thresholds)
attr(qc.lib2, "thresholds")## lower higher
## 434082.9 Inf## lower higher
## 5231.468 Inf# Obtenemos un resumen del número de células
# eliminadas por cada filtro
DataFrame(
LibSize = sum(qc.lib2),
NExprs = sum(qc.nexprs2),
SpikeProp = sum(qc.spike2),
MitoProp = sum(qc.mito2),
Total = sum(discard2)
)## DataFrame with 1 row and 5 columns
## LibSize NExprs SpikeProp MitoProp Total
## <integer> <integer> <integer> <integer> <integer>
## 1 4 0 1 2 62.9.8 Preguntas por resolver
- ¿Cuántos células se descartan con cada filtro?
- ¿Cuántas células se comparten entre ambos filtros?
- ¿Cúal filtro fue más estricto?
- ¿Existen células que el filtro adaptativo excluyera y que no lo hiciera el filtro estricto?
Para contestar estas preguntas podemos emplear:
## discard2
## discard FALSE TRUE Sum
## FALSE 159 0 159
## TRUE 27 6 33
## Sum 186 6 1922.10 ¿Cómo funciona isOutlier()?
Supongamos que la mayor parte del conjunto de datos está formado por células de alta calidad 1
- (Opcional: log-transformar la métrica QC) 1.
- Calcular la mediana de la métrica QC
- Calcular la desviación absoluta de la mediana (MAD2) de la QC
- Identifique los valores atípicos como aquellas celdas con una métrica QC a más de 3 MAD3 de la mediana en la dirección “problemática”.
- Puede controlar cuántas MAD son aceptables
- Puede decidir qué dirección es problemática
QC = Quality Control 1 But we’ll relax that assumption in a few moments 2 MAD is similar to standard deviation 3 Loosely, 3 MADs will retain 99% of non-outliers values that follow a Normal distribution.
Figura explicativa, Diapositiva 24.
2.11 Consideran el Batch
## Determino el bloque (batch) de muestras
batch <- paste0(sce.416b$phenotype, "-", sce.416b$block)
## Versión de isOutlier() que toma en cuenta los bloques de muestras
qc.lib3 <- isOutlier(sce.416b$sum,
log = TRUE,
type = "lower",
batch = batch
)
qc.nexprs3 <- isOutlier(sce.416b$detected,
log = TRUE,
type = "lower",
batch = batch
)
qc.spike3 <- isOutlier(sce.416b$altexps_ERCC_percent,
type = "higher",
batch = batch
)
qc.mito3 <- isOutlier(sce.416b$subsets_Mito_percent,
type = "higher",
batch = batch
)
discard3 <- qc.lib3 | qc.nexprs3 | qc.spike3 | qc.mito3
# Extraemos los límites de valores (thresholds)
attr(qc.lib3, "thresholds")## induced CBFB-MYH11 oncogene expression-20160113 induced CBFB-MYH11 oncogene expression-20160325
## lower 461073.1 399133.7
## higher Inf Inf
## wild type phenotype-20160113 wild type phenotype-20160325
## lower 599794.9 370316.5
## higher Inf Inf# Obtenemos un resumen del número de células
# eliminadas por cada filtro
DataFrame(
LibSize = sum(qc.lib3),
NExprs = sum(qc.nexprs3),
SpikeProp = sum(qc.spike3),
MitoProp = sum(qc.mito3),
Total = sum(discard3)
)## DataFrame with 1 row and 5 columns
## LibSize NExprs SpikeProp MitoProp Total
## <integer> <integer> <integer> <integer> <integer>
## 1 5 4 6 2 92.12 Visualización gráfica de las células de buena calidad
Usaremos el filtro estricto y visualizaremos la distribución de los datos.
# Reducir el nombre en los fenotipos / tratamientos
sce.416b_edited <- sce.416b
sce.416b_edited$phenotype <- ifelse(grepl("induced", sce.416b_edited$phenotype),
"induced", "wild type")
# Verificar
unique(sce.416b_edited$phenotype)## [1] "wild type" "induced"# Agregar columna de genes descartados
sce.416b_edited$discard <- discard
# Visualizacion grafica
plotColData(sce.416b_edited, x="block", y="sum", colour_by="discard",
other_fields="phenotype") + facet_wrap(~phenotype) +
labs(x = "Bloques de secuenciacion", y="Cuentas totales", title = "Numero de cuentas") # Etiquetas en X, Y y titulo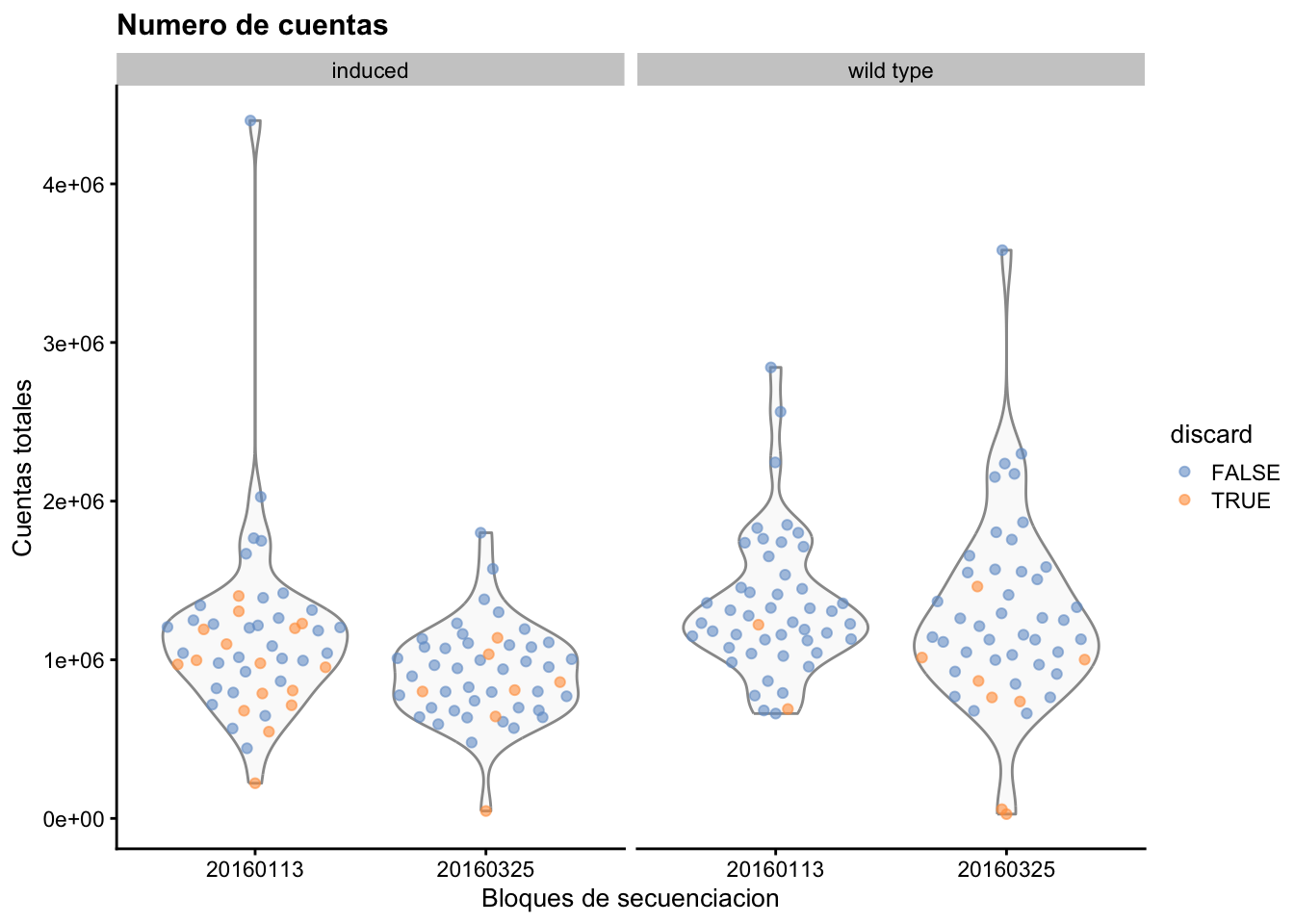
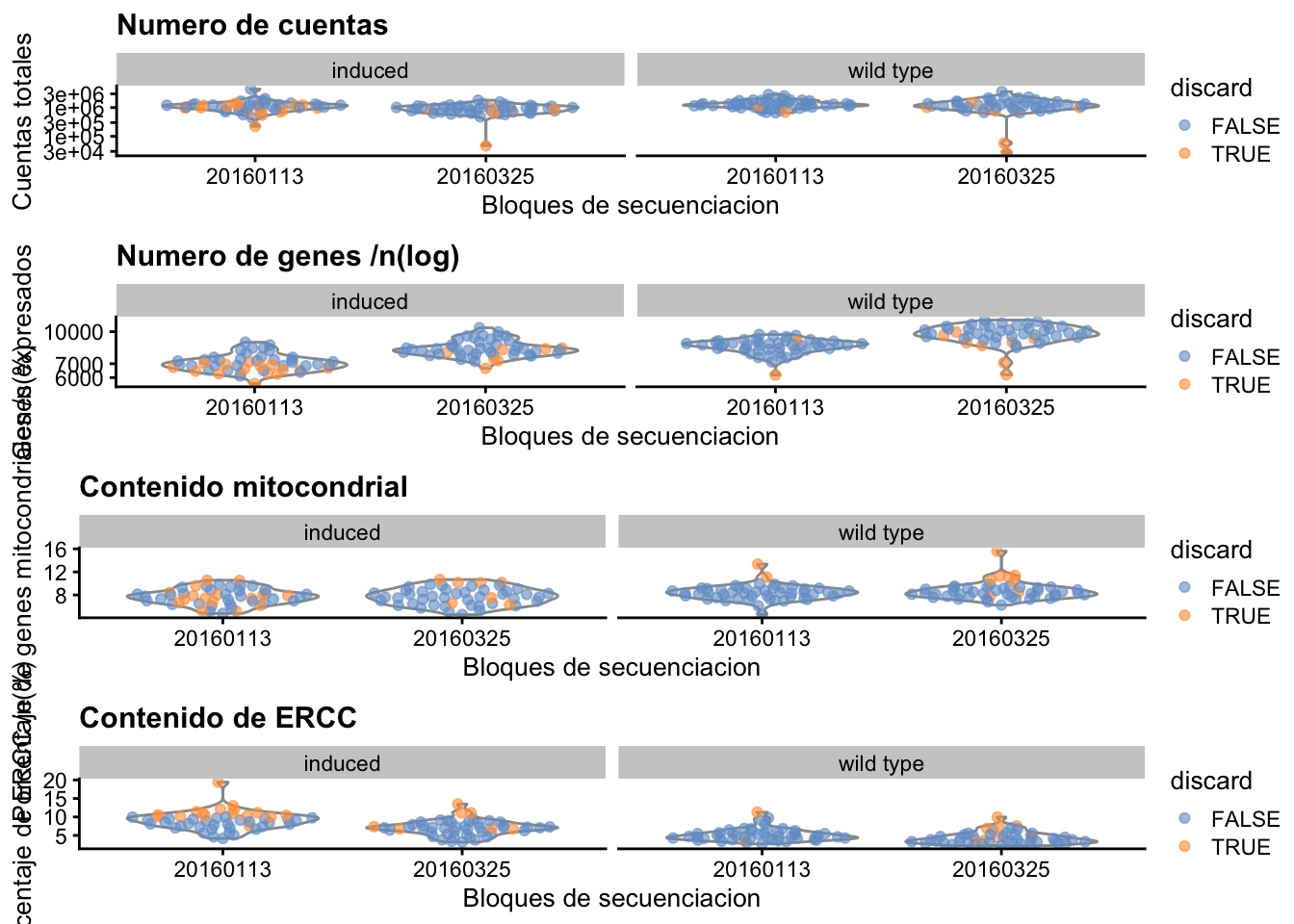
Visualizacion de todas las variables en una sola gráfica.
# Visualizacion grafica
gridExtra::grid.arrange(
plotColData(sce.416b_edited, x="block", y="sum", colour_by="discard",
other_fields="phenotype") + facet_wrap(~phenotype) +
scale_y_log10() + labs(x = "Bloques de secuenciacion", y="Cuentas totales", title = "Numero de cuentas"), # Etiquetas en X, Y y titulo
plotColData(sce.416b_edited, x="block", y="detected", colour_by="discard",
other_fields="phenotype") + facet_wrap(~phenotype) +
scale_y_log10() + labs(x = "Bloques de secuenciacion", y="Genes expresados", title = "Numero de genes /n(log)"), # Etiquetas en X, Y y titulo
plotColData(sce.416b_edited, x="block", y="subsets_Mito_percent",
colour_by="discard", other_fields="phenotype") +
facet_wrap(~phenotype) + labs(x = "Bloques de secuenciacion", y="Porcentaje de genes mitocondriales /n(%)", title = "Contenido mitocondrial"), # Etiquetas en X, Y y titulo
plotColData(sce.416b_edited, x="block", y="altexps_ERCC_percent",
colour_by="discard", other_fields="phenotype") +
facet_wrap(~phenotype) + labs(x = "Bloques de secuenciacion", y="Porcentaje de ERCC /n(%)", title = "Contenido de ERCC"), # Etiquetas en X, Y y titulo,
ncol=1
)
Otra visualización gráfica es:
plotColData(
sce.416b_edited,
x = "sum",
y = "subsets_Mito_percent",
colour_by = "discard",
other_fields = c("block", "phenotype")
) +
facet_grid(block ~ phenotype)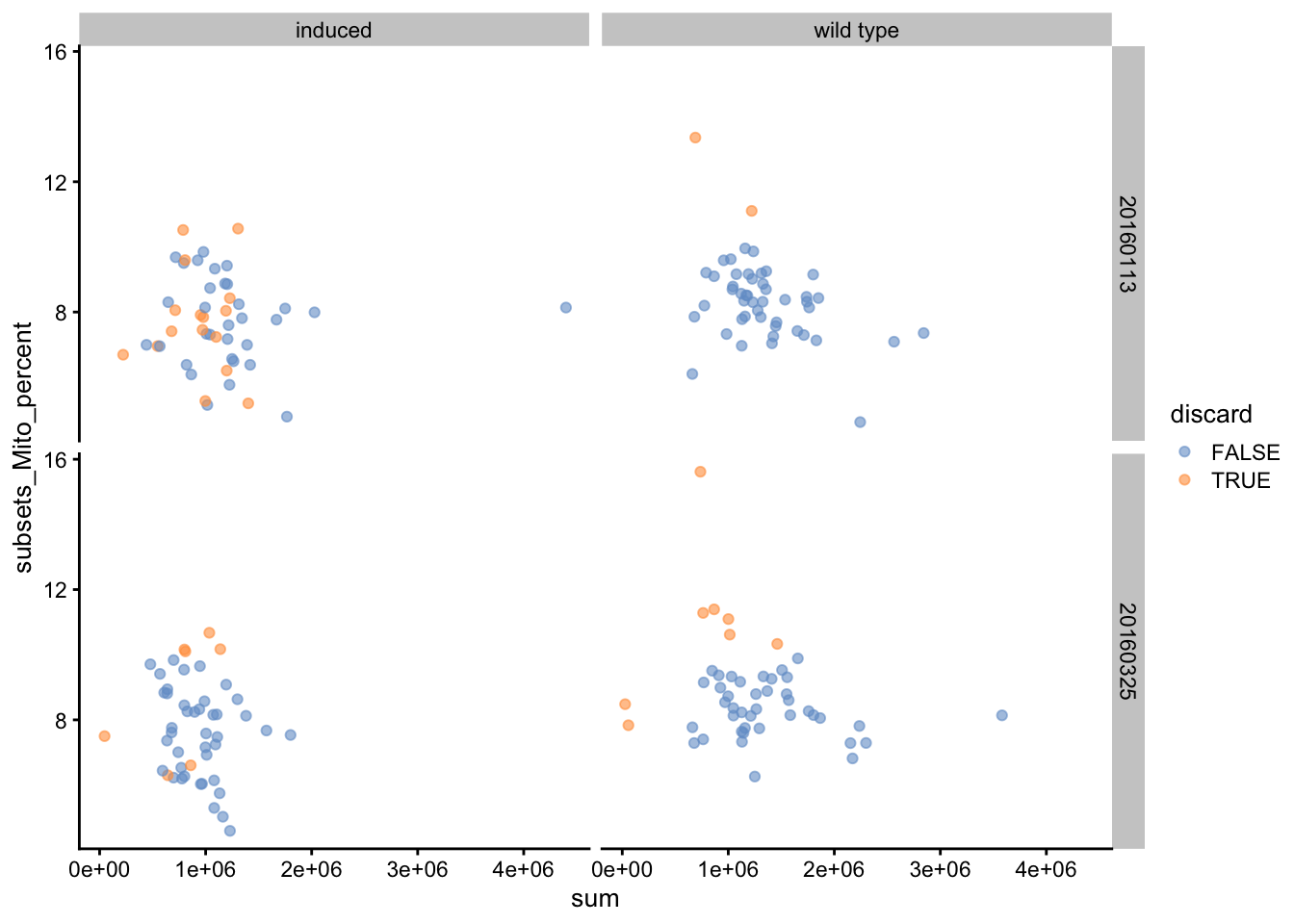
2.13 Identificando droplets vacíos con datos de PBMC
2.13.1 Conceptos básicos
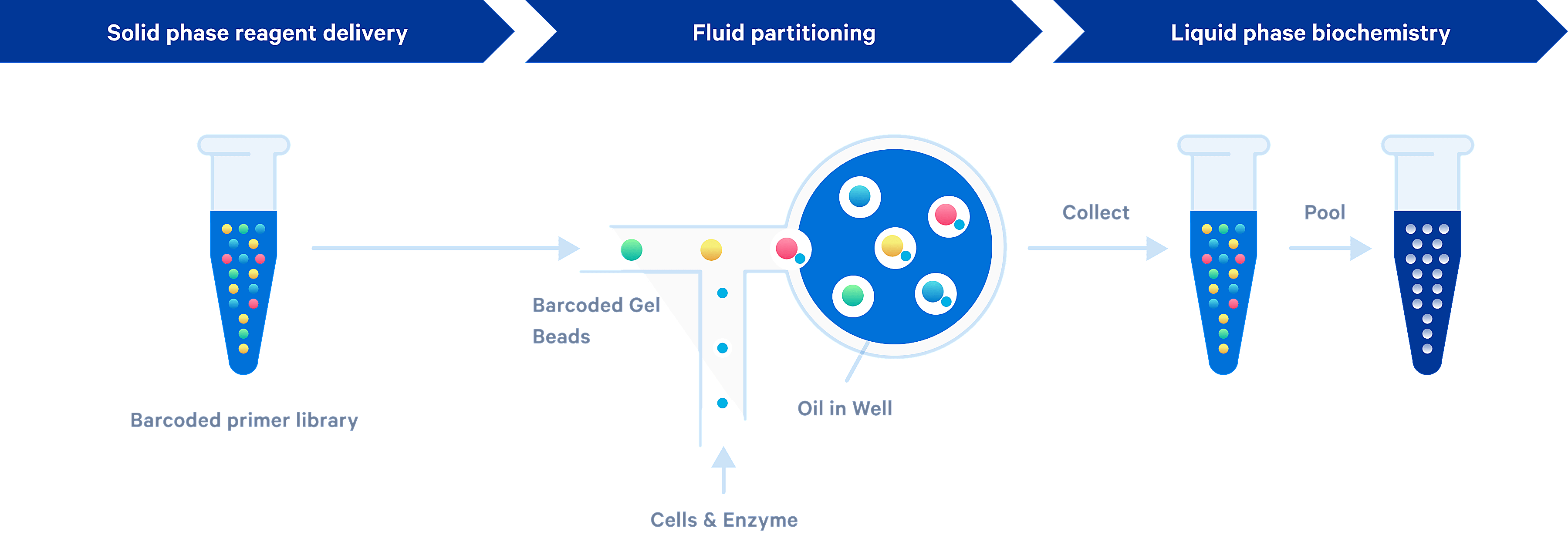
Descripción gráfica de la tecnología Next GEM de 10x Genomics. Fuente: 10x Genomics.
![]()
Opciones algorítmicas para detectar los droplets vacíos. Fuente: Lun et al, Genome Biology, 2019.
2.13.2 Información de los datos
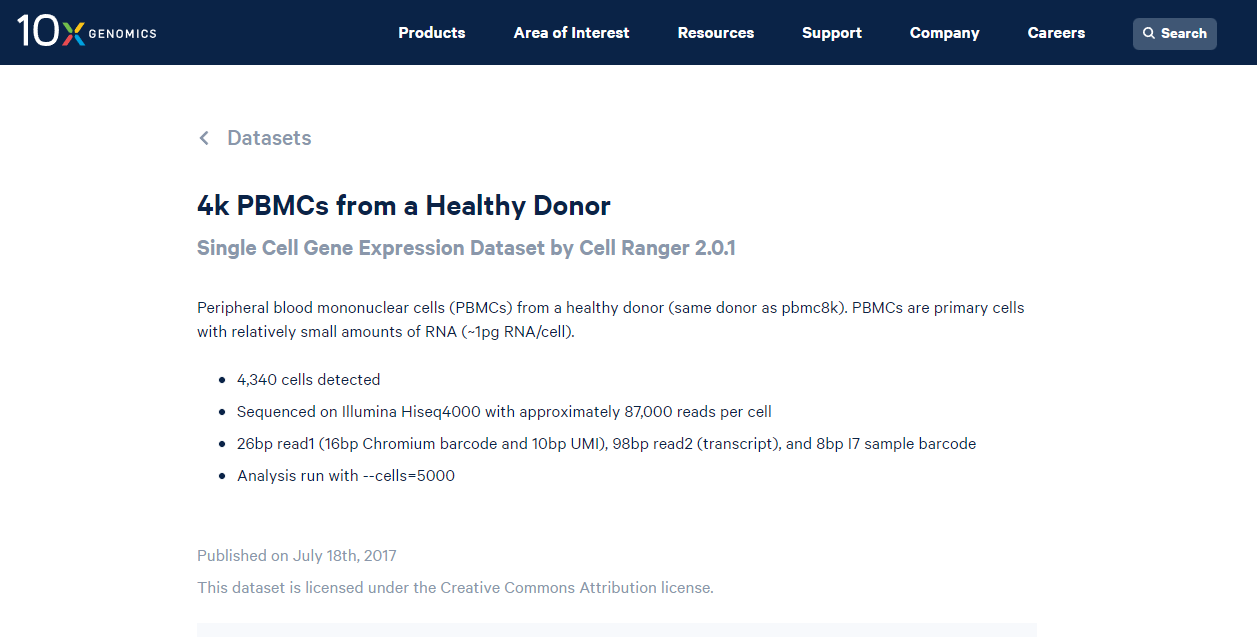
El dataset proviene de células humanas, con un total de 4,340 células con buena calidad. Para más información consulta la página web en 10Xgenomics.
A continuación te presento el reporte de este dataset.
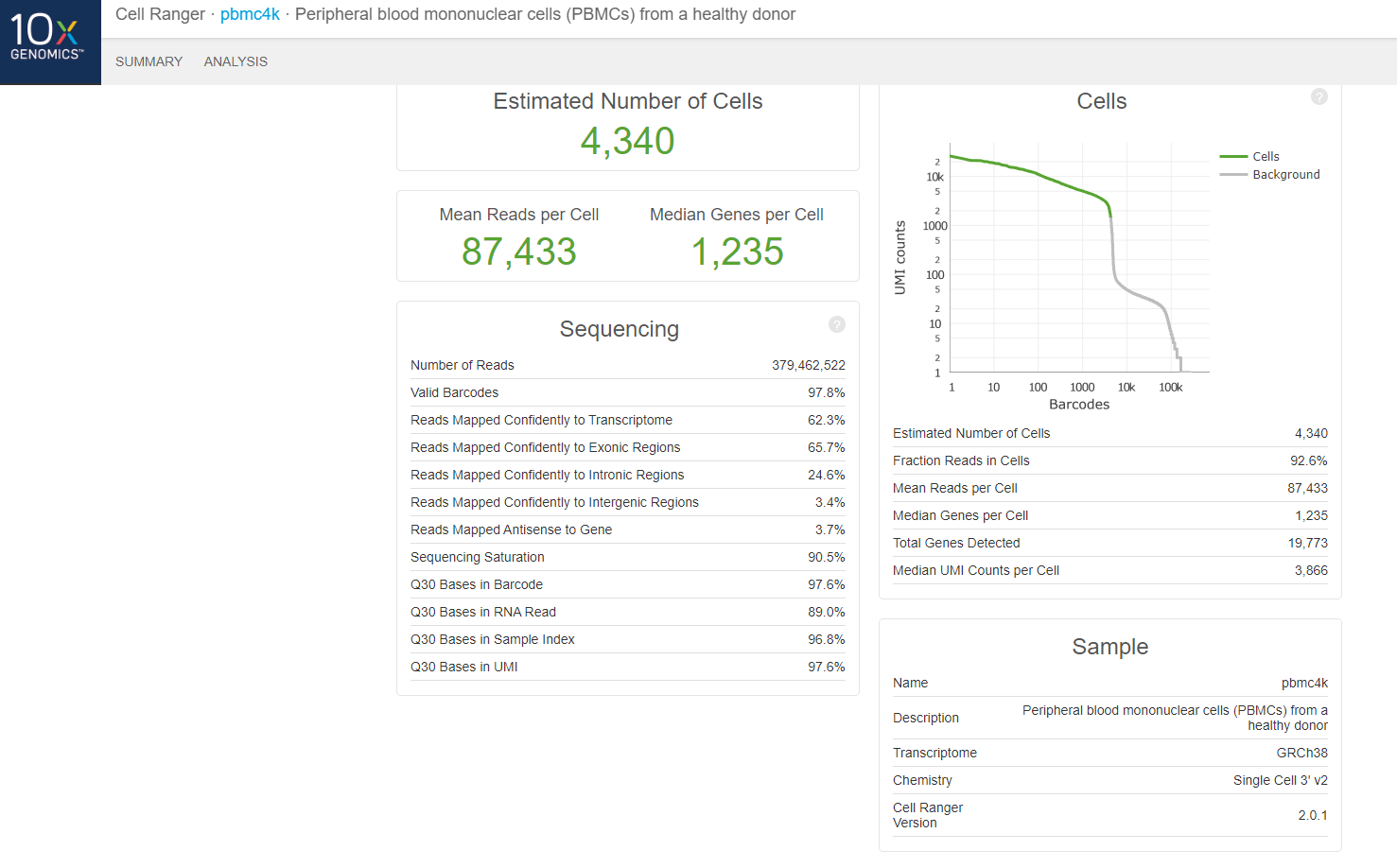
Captura del reporte de secuenciación del dataset.
10Xgenomics realiza un alineamiento genómico con STAR y emplea CellRanger para realizar la limpieza y cuantificación de los datos. Encontrado la información dividia en dos carpetas importantes:
- Gene/cell matrix (raw): Contiene toda la información posterior al alineamiento, sin limpiar. Contiene toda la información, contemplando la presencia de doplets vacíos.
- Gene/cell matrix (filtered): Contiene los datos filtrados por CellRanger.
Dejo a tu consideración la posibilidad de emplear alguno de estos archivos. Ambas carpetas contienen 3 archivos importantes para el análisis de los datos:
barcode.tsv= Secuencia que define a cada célula por identificadores de cada célula.feature.tsv= Genes identificadores, se les midió expresión.matrix.mtx= Unión entre las secuencias y genes.
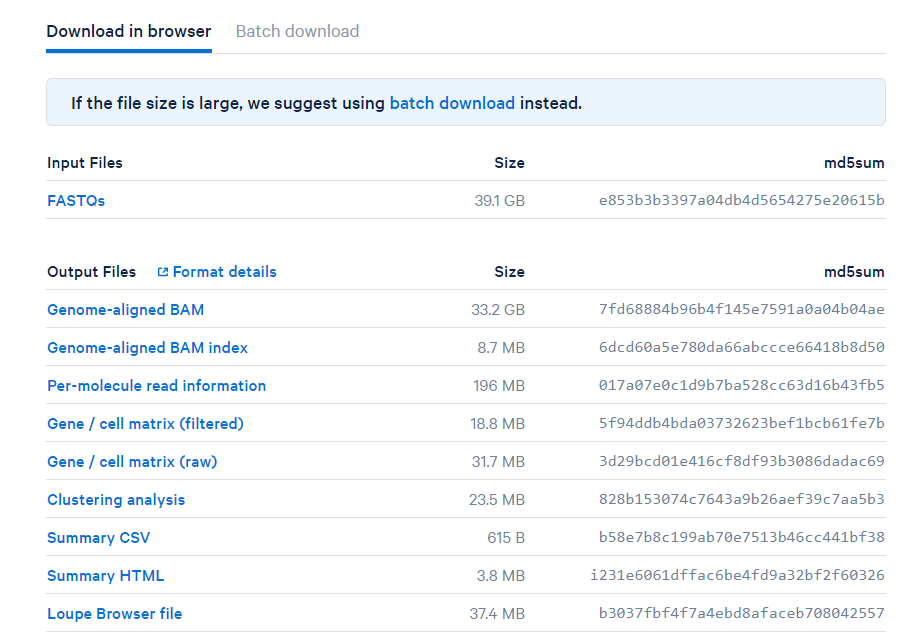
2.13.3 Importar datos en R
Almacenaremos la información de 10Xgenomics en la memoria Cache de la computadora y posteriormente lo almacenaremos en una variable.
# Datos crudos, sin procesar
bfc <- BiocFileCache()
raw.path <- bfcrpath(bfc, file.path(
"http://cf.10xgenomics.com/samples",
"cell-exp/2.1.0/pbmc4k/pbmc4k_raw_gene_bc_matrices.tar.gz"
))
untar(raw.path, exdir = file.path(tempdir(), "pbmc4k"))# Archivos contenidos en Temporales
test_dir <- tempdir()
test_dir <- paste0(test_dir, "/pbmc4k/raw_gene_bc_matrices/GRCh38")
list.files(test_dir) # Encontraremos 3 tipos de archivos: barcodes.tsv, genes.tsv y matrix.mtx ## [1] "barcodes.tsv" "genes.tsv" "matrix.mtx"# Cargar datos
fname <- file.path(tempdir(), "pbmc4k/raw_gene_bc_matrices/GRCh38")
sce.pbmc <- read10xCounts(fname, col.names = TRUE) # Cargar datos de 10Xgenomics de CellRanger
sce.pbmc## class: SingleCellExperiment
## dim: 33694 737280
## metadata(1): Samples
## assays(1): counts
## rownames(33694): ENSG00000243485 ENSG00000237613 ... ENSG00000277475 ENSG00000268674
## rowData names(2): ID Symbol
## colnames(737280): AAACCTGAGAAACCAT-1 AAACCTGAGAAACCGC-1 ... TTTGTCATCTTTAGTC-1 TTTGTCATCTTTCCTC-1
## colData names(2): Sample Barcode
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):Contiene 737280 droplets, aún sin filtrar y evaluando un total de 33,694 genes en este dataset.
Si quisieras descargar estos mismos datos pero filtrados, te dejo la ruta:
# Datos filtrados, sin celulas muertas, sin un alto contenido mitocondrial, sin droplets vacios o con multiples celulas
bfc <- BiocFileCache()
raw.path <- bfcrpath(bfc, file.path(
"http://cf.10xgenomics.com/samples",
"cell-exp/2.1.0/pbmc4k/pbmc4k_filtered_gene_bc_matrices.tar.gz"
))
untar(raw.path, exdir = file.path(tempdir(), "pbmc4k"))2.13.4 Visualización de droplets vacíos
## DataFrame with 737280 rows and 3 columns
## rank total fitted
## <numeric> <integer> <numeric>
## AAACCTGAGAAACCAT-1 219086 1 NA
## AAACCTGAGAAACCGC-1 504862 0 NA
## AAACCTGAGAAACCTA-1 219086 1 NA
## AAACCTGAGAAACGAG-1 504862 0 NA
## AAACCTGAGAAACGCC-1 219086 1 NA
## ... ... ... ...
## TTTGTCATCTTTACAC-1 150232 2 NA
## TTTGTCATCTTTACGT-1 26415 33 NA
## TTTGTCATCTTTAGGG-1 504862 0 NA
## TTTGTCATCTTTAGTC-1 504862 0 NA
## TTTGTCATCTTTCCTC-1 219086 1 NA# Mostremos solo los puntos únicos para acelerar
# el proceso de hacer esta gráfica
uniq <- !duplicated(bcrank$rank)
plot(
bcrank$rank[uniq],
bcrank$total[uniq],
log = "xy",
xlab = "Rank",
ylab = "Total UMI count",
cex.lab = 1.2)## Warning in xy.coords(x, y, xlabel, ylabel, log): 1 y value <= 0 omitted from logarithmic plot# Agregarle los puntos de corte
abline(
h = metadata(bcrank)$inflection,
col = "darkgreen",
lty = 2)
abline(
h = metadata(bcrank)$knee,
col = "dodgerblue",
lty = 2)
legend(
"bottomleft",
legend = c("Inflection", "Knee"),
col = c("darkgreen", "dodgerblue"),
lty = 2,
cex = 1.2)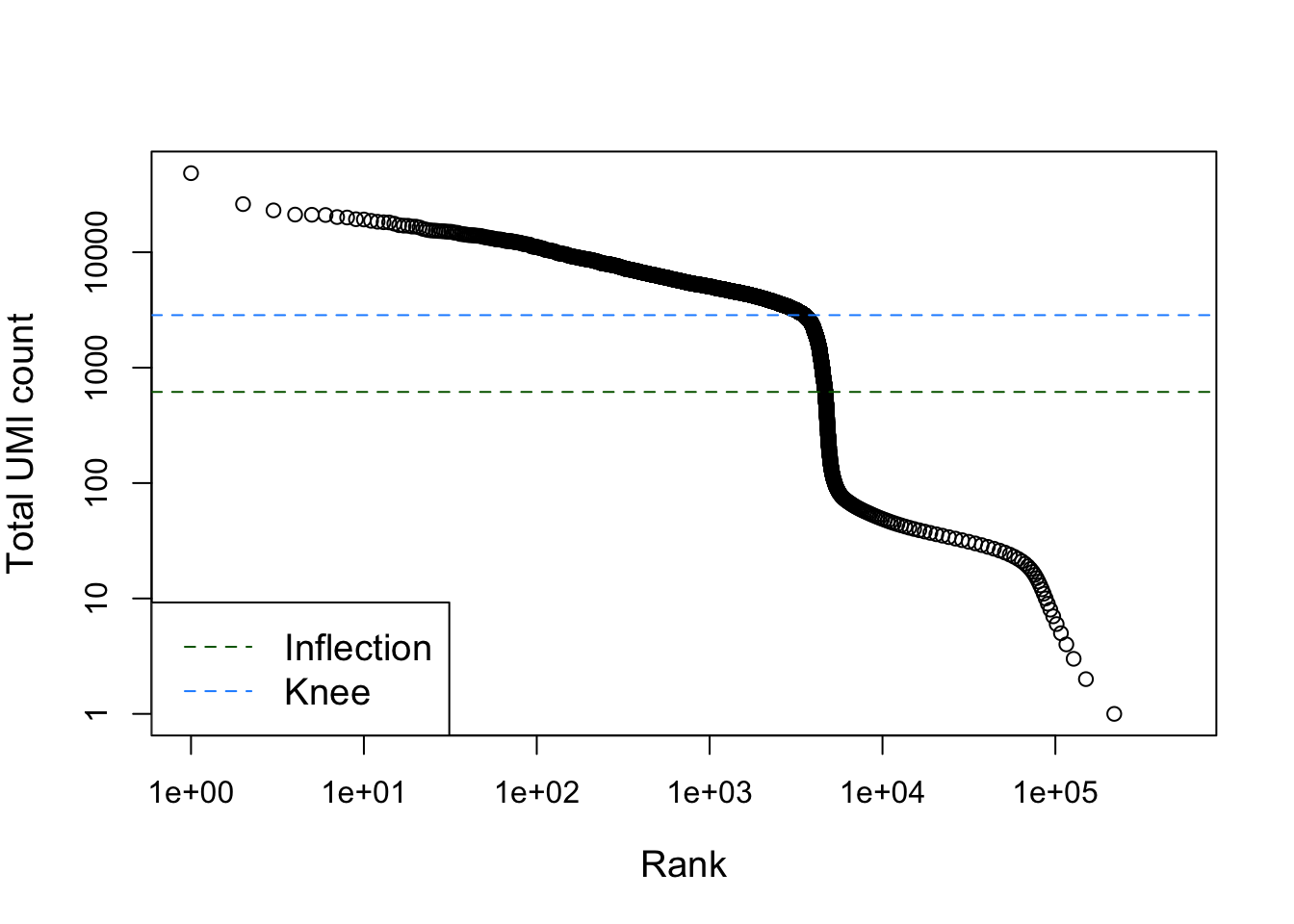
UMI = Secuencia única de cada unión en la bead (unique molecular identifiers).
Encontremos los droplets vacíos usando emptyDrops(). Los siguientes pasos demoran un tiempo.
## Usemos DropletUtils para encontrar los droplets
set.seed(100)
e.out <- emptyDrops(counts(sce.pbmc))
# Revisa ?emptyDrops para una explicación de por qué hay valores NA
summary(e.out$FDR <= 0.001)## Mode FALSE TRUE NA's
## logical 1006 4283 731991set.seed(100)
limit <- 100
all.out <- emptyDrops(counts(sce.pbmc), lower = limit, test.ambient = TRUE)
# Idealmente, este histograma debería verse uniforme.
# Picos grandes cerca de cero indican que los _barcodes_
# con un número total de cuentas menor a "lower" no son
# de origen ambiental.
hist(all.out$PValue[all.out$Total <= limit &
all.out$Total > 0],
xlab = "P-value",
main = "",
col = "grey80")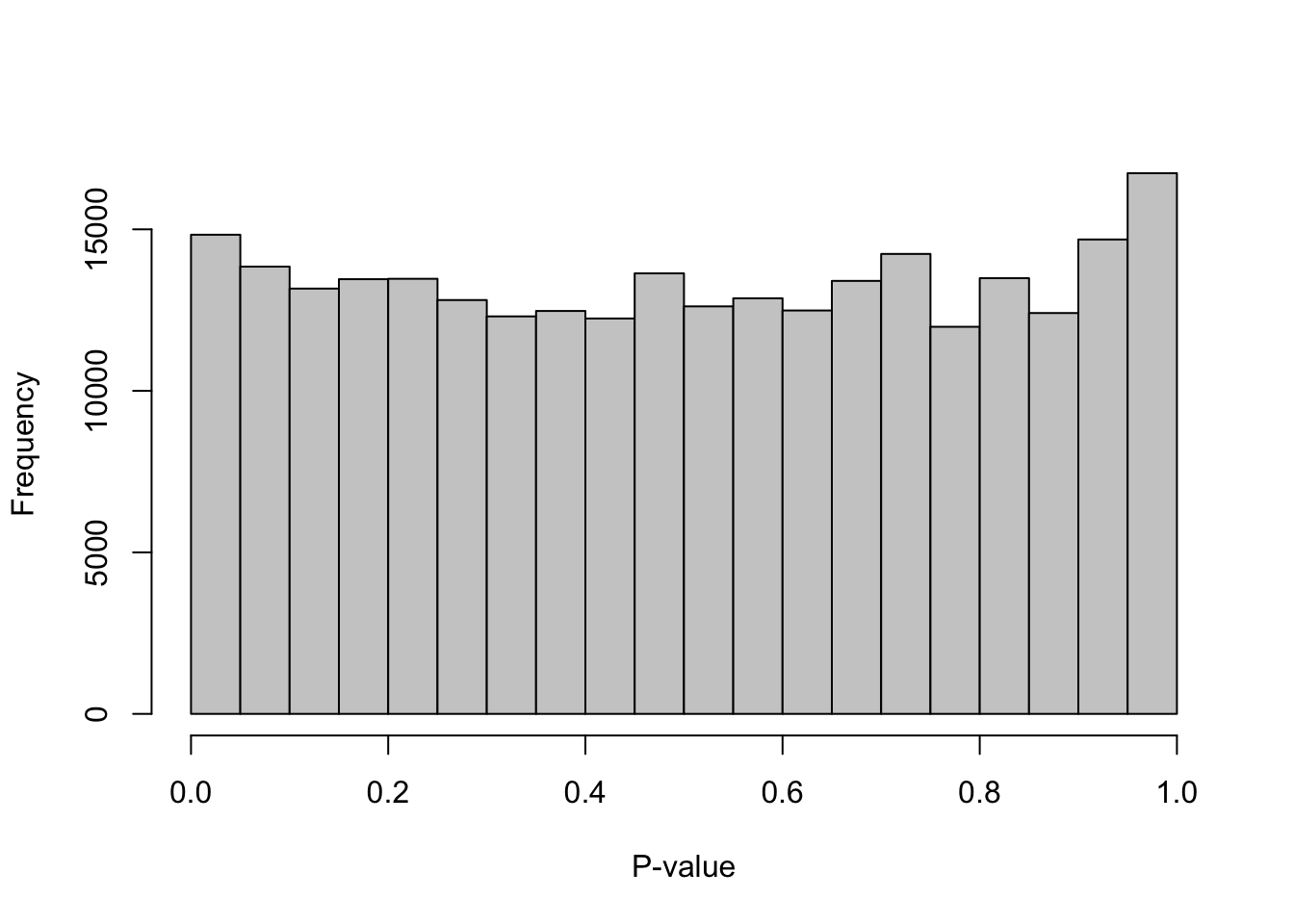
2.13.5 Anotación de genes y eliminación de doples vacíos
# Anotación de los genes
rownames(sce.pbmc) <- uniquifyFeatureNames(
rowData(sce.pbmc)$ID, rowData(sce.pbmc)$Symbol
)
location <- mapIds(EnsDb.Hsapiens.v86,
keys = rowData(sce.pbmc)$ID,
column = "SEQNAME", keytype = "GENEID"
)## Warning: Unable to map 144 of 33694 requested IDs.2.13.6 Control de calidad
# Obtener las estadisticas en un archivo aparte
stats <- perCellQCMetrics(sce.pbmc,
subsets = list(Mito = which(location == "MT"))
)
high.mito <- isOutlier(stats$subsets_Mito_percent,
type = "higher"
)
# Eliminar secuencias con alto contenido de genes mitocondriales
sce.pbmc <- sce.pbmc[, !high.mito]2.15 Detalles de la sesión de R
## [1] "2023-08-10 10:08:46 EDT"## user system elapsed
## 1743.470 121.432 136359.256## ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
## setting value
## version R version 4.3.1 (2023-06-16)
## os macOS Ventura 13.4.1
## system aarch64, darwin20
## ui RStudio
## language (EN)
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz America/New_York
## date 2023-08-10
## rstudio 2023.06.0+421 Mountain Hydrangea (desktop)
## pandoc 3.1.1 @ /Applications/RStudio.app/Contents/Resources/app/quarto/bin/tools/ (via rmarkdown)
##
## ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
## package * version date (UTC) lib source
## abind 1.4-5 2016-07-21 [1] CRAN (R 4.3.0)
## AnnotationDbi * 1.62.2 2023-07-02 [1] Bioconductor
## AnnotationFilter * 1.24.0 2023-05-08 [1] Bioconductor
## AnnotationHub * 3.8.0 2023-05-08 [1] Bioconductor
## beachmat 2.16.0 2023-05-08 [1] Bioconductor
## beeswarm 0.4.0 2021-06-01 [1] CRAN (R 4.3.0)
## Biobase * 2.60.0 2023-05-08 [1] Bioconductor
## BiocFileCache * 2.8.0 2023-05-08 [1] Bioconductor
## BiocGenerics * 0.46.0 2023-06-04 [1] Bioconductor
## BiocIO 1.10.0 2023-05-08 [1] Bioconductor
## BiocManager 1.30.21.1 2023-07-18 [1] CRAN (R 4.3.0)
## BiocNeighbors 1.18.0 2023-05-08 [1] Bioconductor
## BiocParallel 1.34.2 2023-05-28 [1] Bioconductor
## BiocSingular 1.16.0 2023-05-08 [1] Bioconductor
## BiocVersion 3.17.1 2022-12-20 [1] Bioconductor
## biomaRt 2.56.1 2023-06-11 [1] Bioconductor
## Biostrings 2.68.1 2023-05-21 [1] Bioconductor
## bit 4.0.5 2022-11-15 [1] CRAN (R 4.3.0)
## bit64 4.0.5 2020-08-30 [1] CRAN (R 4.3.0)
## bitops 1.0-7 2021-04-24 [1] CRAN (R 4.3.0)
## blob 1.2.4 2023-03-17 [1] CRAN (R 4.3.0)
## bluster * 1.10.0 2023-05-08 [1] Bioconductor
## bookdown 0.34 2023-05-09 [1] CRAN (R 4.3.0)
## bslib 0.5.0 2023-06-09 [1] CRAN (R 4.3.0)
## cachem 1.0.8 2023-05-01 [1] CRAN (R 4.3.0)
## cli 3.6.1 2023-03-23 [1] CRAN (R 4.3.0)
## cluster 2.1.4 2022-08-22 [1] CRAN (R 4.3.1)
## codetools 0.2-19 2023-02-01 [1] CRAN (R 4.3.1)
## colorspace 2.1-0 2023-01-23 [1] CRAN (R 4.3.0)
## cowplot 1.1.1 2020-12-30 [1] CRAN (R 4.3.0)
## crayon 1.5.2 2022-09-29 [1] CRAN (R 4.3.0)
## curl 5.0.1 2023-06-07 [1] CRAN (R 4.3.0)
## DBI 1.1.3 2022-06-18 [1] CRAN (R 4.3.0)
## dbplyr * 2.3.3 2023-07-07 [1] CRAN (R 4.3.0)
## DelayedArray 0.26.7 2023-07-28 [1] Bioconductor
## DelayedMatrixStats 1.22.1 2023-06-09 [1] Bioconductor
## digest 0.6.33 2023-07-07 [1] CRAN (R 4.3.0)
## dplyr * 1.1.2 2023-04-20 [1] CRAN (R 4.3.0)
## dqrng 0.3.0 2021-05-01 [1] CRAN (R 4.3.0)
## DropletUtils * 1.20.0 2023-05-08 [1] Bioconductor
## edgeR 3.42.4 2023-06-04 [1] Bioconductor
## ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.3.0)
## EnsDb.Hsapiens.v86 * 2.99.0 2023-07-29 [1] Bioconductor
## ensembldb * 2.24.0 2023-05-08 [1] Bioconductor
## evaluate 0.21 2023-05-05 [1] CRAN (R 4.3.0)
## ExperimentHub 2.8.1 2023-07-16 [1] Bioconductor
## fansi 1.0.4 2023-01-22 [1] CRAN (R 4.3.0)
## farver 2.1.1 2022-07-06 [1] CRAN (R 4.3.0)
## fastmap 1.1.1 2023-02-24 [1] CRAN (R 4.3.0)
## filelock 1.0.2 2018-10-05 [1] CRAN (R 4.3.0)
## FNN 1.1.3.2 2023-03-20 [1] CRAN (R 4.3.0)
## generics 0.1.3 2022-07-05 [1] CRAN (R 4.3.0)
## GenomeInfoDb * 1.36.1 2023-07-02 [1] Bioconductor
## GenomeInfoDbData 1.2.10 2023-06-08 [1] Bioconductor
## GenomicAlignments 1.36.0 2023-05-08 [1] Bioconductor
## GenomicFeatures * 1.52.1 2023-07-02 [1] Bioconductor
## GenomicRanges * 1.52.0 2023-05-08 [1] Bioconductor
## ggbeeswarm 0.7.2 2023-04-29 [1] CRAN (R 4.3.0)
## ggplot2 * 3.4.2 2023-04-03 [1] CRAN (R 4.3.0)
## ggrepel * 0.9.3 2023-02-03 [1] CRAN (R 4.3.0)
## glue 1.6.2 2022-02-24 [1] CRAN (R 4.3.0)
## gridExtra 2.3 2017-09-09 [1] CRAN (R 4.3.0)
## gtable 0.3.3 2023-03-21 [1] CRAN (R 4.3.0)
## HDF5Array 1.28.1 2023-05-08 [1] Bioconductor
## here 1.0.1 2020-12-13 [1] CRAN (R 4.3.0)
## highr 0.10 2022-12-22 [1] CRAN (R 4.3.0)
## hms 1.1.3 2023-03-21 [1] CRAN (R 4.3.0)
## htmltools 0.5.5 2023-03-23 [1] CRAN (R 4.3.0)
## httpuv 1.6.11 2023-05-11 [1] CRAN (R 4.3.0)
## httr 1.4.6 2023-05-08 [1] CRAN (R 4.3.0)
## igraph 1.5.0.1 2023-07-23 [1] CRAN (R 4.3.0)
## interactiveDisplayBase 1.38.0 2023-05-08 [1] Bioconductor
## IRanges * 2.34.1 2023-07-02 [1] Bioconductor
## irlba 2.3.5.1 2022-10-03 [1] CRAN (R 4.3.0)
## jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.3.0)
## jsonlite 1.8.7 2023-06-29 [1] CRAN (R 4.3.0)
## kableExtra * 1.3.4 2021-02-20 [1] CRAN (R 4.3.0)
## KEGGREST 1.40.0 2023-05-08 [1] Bioconductor
## knitr 1.43 2023-05-25 [1] CRAN (R 4.3.0)
## labeling 0.4.2 2020-10-20 [1] CRAN (R 4.3.0)
## later 1.3.1 2023-05-02 [1] CRAN (R 4.3.0)
## lattice 0.21-8 2023-04-05 [1] CRAN (R 4.3.1)
## lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.3.0)
## lifecycle 1.0.3 2022-10-07 [1] CRAN (R 4.3.0)
## limma 3.56.2 2023-06-04 [1] Bioconductor
## locfit 1.5-9.8 2023-06-11 [1] CRAN (R 4.3.0)
## magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.3.0)
## Matrix * 1.6-0 2023-07-08 [1] CRAN (R 4.3.0)
## MatrixGenerics * 1.12.3 2023-07-30 [1] Bioconductor
## matrixStats * 1.0.0 2023-06-02 [1] CRAN (R 4.3.0)
## memoise 2.0.1 2021-11-26 [1] CRAN (R 4.3.0)
## metapod 1.8.0 2023-04-25 [1] Bioconductor
## mime 0.12 2021-09-28 [1] CRAN (R 4.3.0)
## munsell 0.5.0 2018-06-12 [1] CRAN (R 4.3.0)
## patchwork * 1.1.2 2022-08-19 [1] CRAN (R 4.3.0)
## PCAtools * 2.12.0 2023-05-08 [1] Bioconductor
## pheatmap * 1.0.12 2019-01-04 [1] CRAN (R 4.3.0)
## pillar 1.9.0 2023-03-22 [1] CRAN (R 4.3.0)
## pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.3.0)
## plyr 1.8.8 2022-11-11 [1] CRAN (R 4.3.0)
## png 0.1-8 2022-11-29 [1] CRAN (R 4.3.0)
## prettyunits 1.1.1 2020-01-24 [1] CRAN (R 4.3.0)
## progress 1.2.2 2019-05-16 [1] CRAN (R 4.3.0)
## promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.3.0)
## ProtGenerics 1.32.0 2023-05-08 [1] Bioconductor
## purrr 1.0.1 2023-01-10 [1] CRAN (R 4.3.0)
## R.methodsS3 1.8.2 2022-06-13 [1] CRAN (R 4.3.0)
## R.oo 1.25.0 2022-06-12 [1] CRAN (R 4.3.0)
## R.utils 2.12.2 2022-11-11 [1] CRAN (R 4.3.0)
## R6 2.5.1 2021-08-19 [1] CRAN (R 4.3.0)
## rappdirs 0.3.3 2021-01-31 [1] CRAN (R 4.3.0)
## RColorBrewer 1.1-3 2022-04-03 [1] CRAN (R 4.3.0)
## Rcpp 1.0.11 2023-07-06 [1] CRAN (R 4.3.0)
## RCurl 1.98-1.12 2023-03-27 [1] CRAN (R 4.3.0)
## reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.3.0)
## restfulr 0.0.15 2022-06-16 [1] CRAN (R 4.3.0)
## rhdf5 2.44.0 2023-05-08 [1] Bioconductor
## rhdf5filters 1.12.1 2023-05-08 [1] Bioconductor
## Rhdf5lib 1.22.0 2023-05-08 [1] Bioconductor
## rjson 0.2.21 2022-01-09 [1] CRAN (R 4.3.0)
## rlang 1.1.1 2023-04-28 [1] CRAN (R 4.3.0)
## rmarkdown 2.23 2023-07-01 [1] CRAN (R 4.3.0)
## rprojroot 2.0.3 2022-04-02 [1] CRAN (R 4.3.0)
## Rsamtools 2.16.0 2023-06-04 [1] Bioconductor
## RSQLite 2.3.1 2023-04-03 [1] CRAN (R 4.3.0)
## rstudioapi 0.15.0 2023-07-07 [1] CRAN (R 4.3.0)
## rsvd 1.0.5 2021-04-16 [1] CRAN (R 4.3.0)
## rtracklayer 1.60.0 2023-05-08 [1] Bioconductor
## Rtsne 0.16 2022-04-17 [1] CRAN (R 4.3.0)
## rvest 1.0.3 2022-08-19 [1] CRAN (R 4.3.0)
## S4Arrays 1.0.5 2023-07-24 [1] Bioconductor
## S4Vectors * 0.38.1 2023-05-08 [1] Bioconductor
## sass 0.4.7 2023-07-15 [1] CRAN (R 4.3.0)
## ScaledMatrix 1.8.1 2023-05-08 [1] Bioconductor
## scales 1.2.1 2022-08-20 [1] CRAN (R 4.3.0)
## scater * 1.28.0 2023-04-25 [1] Bioconductor
## scran * 1.28.2 2023-07-23 [1] Bioconductor
## scRNAseq * 2.14.0 2023-04-27 [1] Bioconductor
## scuttle * 1.9.4 2023-01-23 [1] Bioconductor
## sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.3.0)
## shiny 1.7.4.1 2023-07-06 [1] CRAN (R 4.3.0)
## SingleCellExperiment * 1.22.0 2023-05-08 [1] Bioconductor
## sparseMatrixStats 1.12.2 2023-07-02 [1] Bioconductor
## statmod 1.5.0 2023-01-06 [1] CRAN (R 4.3.0)
## stringi 1.7.12 2023-01-11 [1] CRAN (R 4.3.0)
## stringr 1.5.0 2022-12-02 [1] CRAN (R 4.3.0)
## SummarizedExperiment * 1.30.2 2023-06-06 [1] Bioconductor
## svglite 2.1.1 2023-01-10 [1] CRAN (R 4.3.0)
## systemfonts 1.0.4 2022-02-11 [1] CRAN (R 4.3.0)
## tibble 3.2.1 2023-03-20 [1] CRAN (R 4.3.0)
## tidyselect 1.2.0 2022-10-10 [1] CRAN (R 4.3.0)
## utf8 1.2.3 2023-01-31 [1] CRAN (R 4.3.0)
## uwot 0.1.16 2023-06-29 [1] CRAN (R 4.3.0)
## vctrs 0.6.3 2023-06-14 [1] CRAN (R 4.3.0)
## vipor 0.4.5 2017-03-22 [1] CRAN (R 4.3.0)
## viridis 0.6.4 2023-07-22 [1] CRAN (R 4.3.0)
## viridisLite 0.4.2 2023-05-02 [1] CRAN (R 4.3.0)
## webshot 0.5.5 2023-06-26 [1] CRAN (R 4.3.0)
## withr 2.5.0 2022-03-03 [1] CRAN (R 4.3.0)
## xfun 0.39 2023-04-20 [1] CRAN (R 4.3.0)
## XML 3.99-0.14 2023-03-19 [1] CRAN (R 4.3.0)
## xml2 1.3.5 2023-07-06 [1] CRAN (R 4.3.0)
## xtable 1.8-4 2019-04-21 [1] CRAN (R 4.3.0)
## XVector 0.40.0 2023-05-08 [1] Bioconductor
## yaml 2.3.7 2023-01-23 [1] CRAN (R 4.3.0)
## zlibbioc 1.46.0 2023-05-08 [1] Bioconductor
##
## [1] /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/library
##
## ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────