7 Identificación de genes marcadores
Instructora: Yalbi I. Balderas-Martínez.
7.1 Diapositivas de Peter Hickey
Ver las diapositivas originales aquí
7.2 Motivación
Ahora que hemos obtenido los clústeres, nos preguntamos, pero qué son? (e.g. ¿qué tipo celular es el clúster 1?)
¿Cuáles genes están dirigiendo el agrupamiento (e.g., ¿cuáles son los genes diferencialmente expresados entre los clústeres 1 y 2?)
Idea: Mirar las diferencias en los perfiles de expresión de las células de los diferentes clústeres
7.3 Dataset ilustrativo: PBMC4k 10X sin filtrar
# Usemos datos de pbmc4k
library(BiocFileCache)
bfc <- BiocFileCache()
raw.path <- bfcrpath(bfc, file.path(
"http://cf.10xgenomics.com/samples",
"cell-exp/2.1.0/pbmc4k/pbmc4k_raw_gene_bc_matrices.tar.gz"
))
untar(raw.path, exdir = file.path(tempdir(), "pbmc4k"))library(DropletUtils)
library(Matrix)
fname <- file.path(tempdir(), "pbmc4k/raw_gene_bc_matrices/GRCh38")
sce.pbmc <- read10xCounts(fname, col.names = TRUE) # 737280 colsDataset “Células mononucleares humanas de sangre periférica” de 10X Genomics (Zheng, G.X.Y. et al, 2017) 5
7.3.1 Anotación
# Anotación de los genes
library(scater)
rownames(sce.pbmc) <- uniquifyFeatureNames(
rowData(sce.pbmc)$ID, rowData(sce.pbmc)$Symbol
)
library(EnsDb.Hsapiens.v86)
rowData(sce.pbmc)$location <- mapIds(EnsDb.Hsapiens.v86,
keys = rowData(sce.pbmc)$ID,
column = "SEQNAME", keytype = "GENEID"
)
# Information on the chromosome location of the genes
rowData(sce.pbmc)$location[is.na(rowData(sce.pbmc)$location)] <- "NA"
rowData(sce.pbmc)## DataFrame with 33694 rows and 3 columns
## ID Symbol location
## <character> <character> <character>
## RP11-34P13.3 ENSG00000243485 RP11-34P13.3 1
## FAM138A ENSG00000237613 FAM138A 1
## OR4F5 ENSG00000186092 OR4F5 1
## RP11-34P13.7 ENSG00000238009 RP11-34P13.7 1
## RP11-34P13.8 ENSG00000239945 RP11-34P13.8 1
## ... ... ... ...
## AC233755.2 ENSG00000277856 AC233755.2 KI270726.1
## AC233755.1 ENSG00000275063 AC233755.1 KI270726.1
## AC240274.1 ENSG00000271254 AC240274.1 KI270711.1
## AC213203.1 ENSG00000277475 AC213203.1 KI270713.1
## FAM231B ENSG00000268674 FAM231B KI270713.1## DataFrame with 3125 rows and 3 columns
## ID Symbol location
## <character> <character> <character>
## RP11-34P13.3 ENSG00000243485 RP11-34P13.3 1
## FAM138A ENSG00000237613 FAM138A 1
## OR4F5 ENSG00000186092 OR4F5 1
## RP11-34P13.7 ENSG00000238009 RP11-34P13.7 1
## RP11-34P13.8 ENSG00000239945 RP11-34P13.8 1
## ... ... ... ...
## SH3BP5L ENSG00000175137 SH3BP5L 1
## ZNF672 ENSG00000171161 ZNF672 1
## ZNF692 ENSG00000171163 ZNF692 1
## AL672294.1 ENSG00000227237 AL672294.1 1
## PGBD2 ENSG00000185220 PGBD2 1# 33694 rows y 737280 cols
# Detección de células
set.seed(100)
e.out <- emptyDrops(counts(sce.pbmc))
summary(e.out$FDR <= 0.001) # La mayoría se fue porque las que tienen menos de 100 se consideraron vacías## Mode FALSE TRUE NA's
## logical 1006 4283 7319917.4 Motivación - continuación
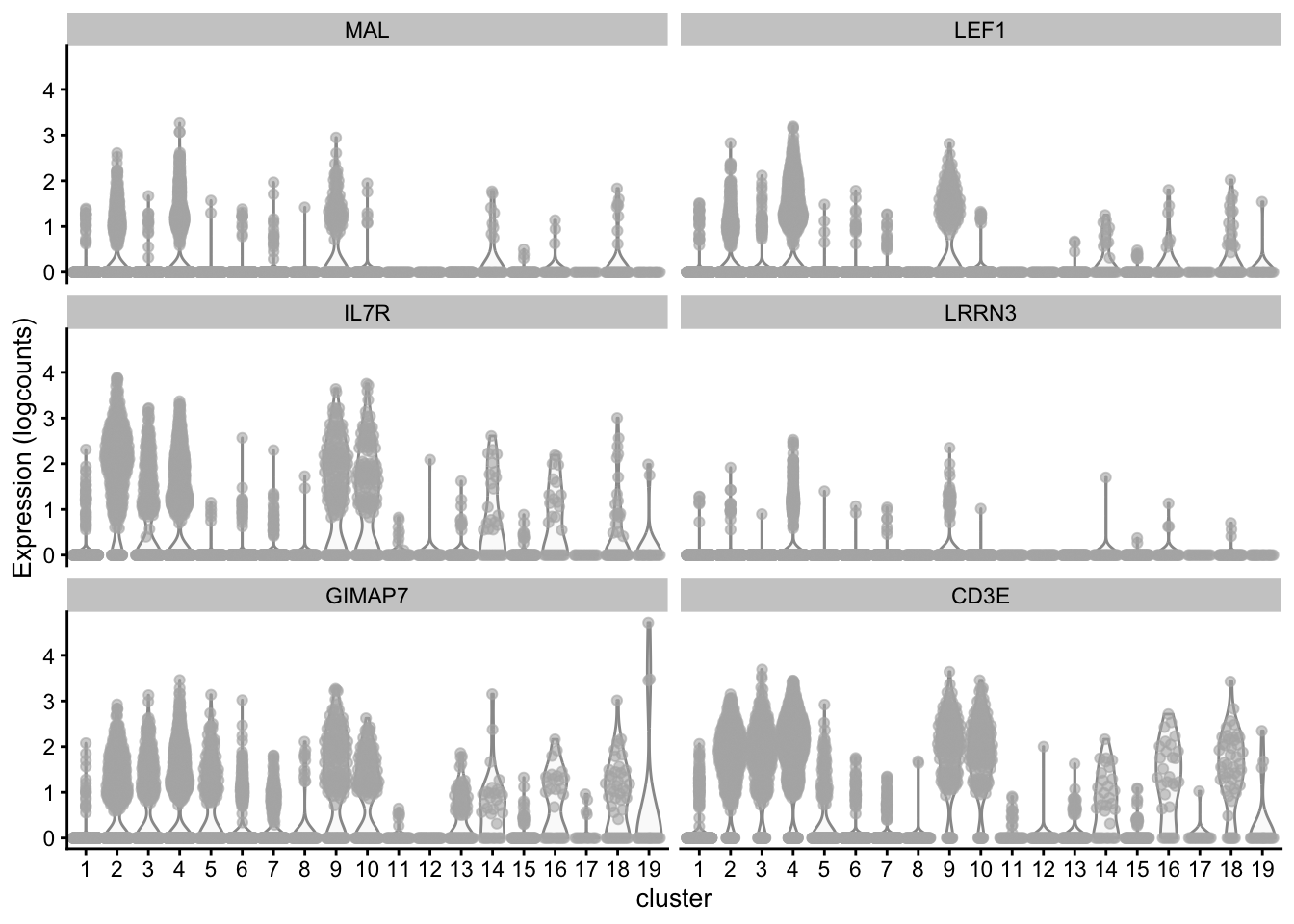
¿Algunos de estos genes están asociados con los resultados de clustering?
# El gen 1 está asociado con el clustering?
plotExpression(sce.pbmc,
features = rownames(sce.pbmc)[1],
x = "cluster", colour_by = "cluster"
)
# El gen 2 está asociado con el clustering?
plotExpression(sce.pbmc,
features = rownames(sce.pbmc)[2],
x = "cluster", colour_by = "cluster"
)
# El gen 2512 está asociado con el clustering?
plotExpression(sce.pbmc,
features = rownames(sce.pbmc)[2512],
x = "cluster", colour_by = "cluster"
)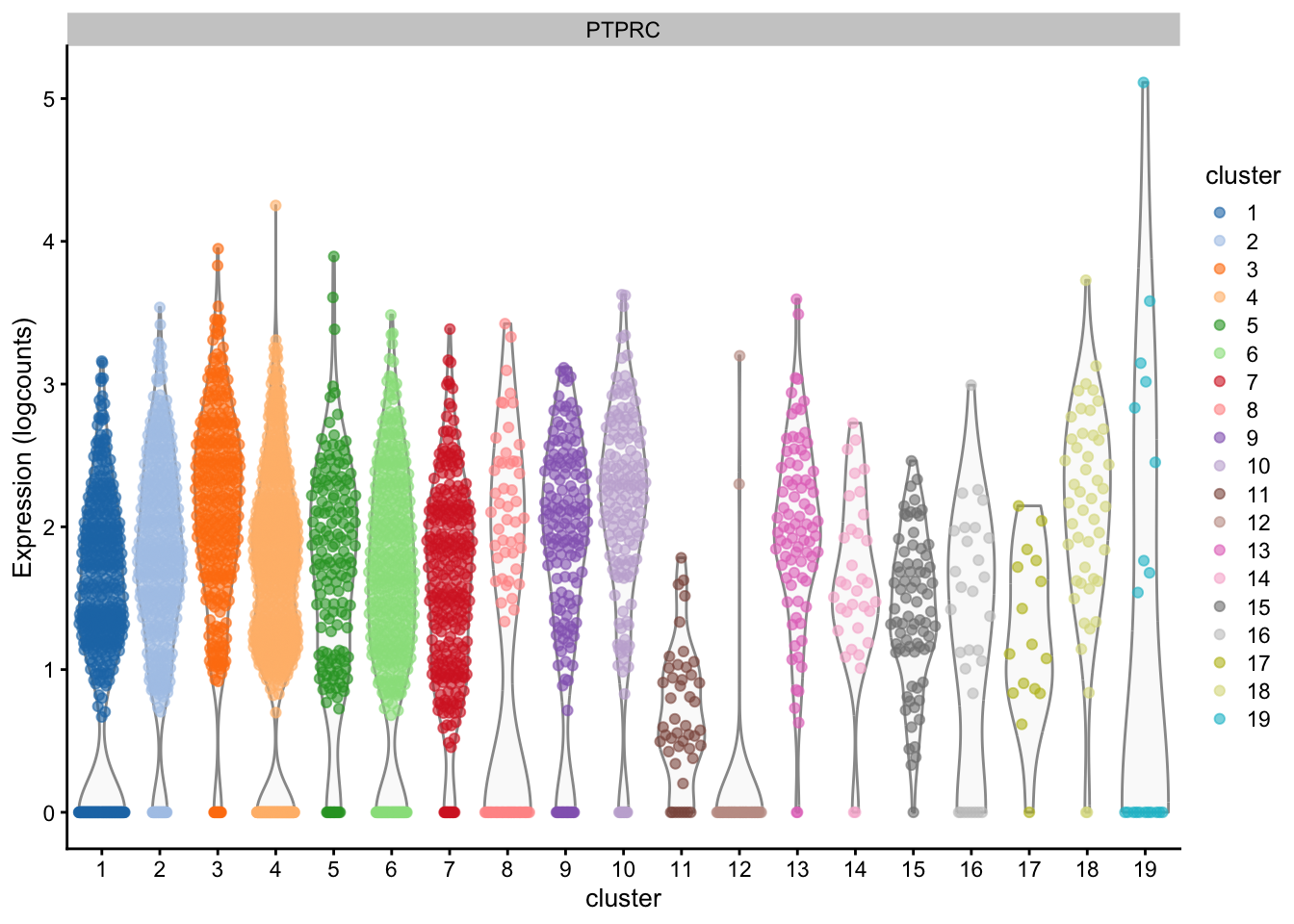
# PTPRC también es conocido como CD45 que antes se llamaba leukocyte common antigen (LCA). Esto sugiere que casi todos los clusters son leucocitos (excepto el 9)# El gen CD3E está asociado con el clustering?
plotExpression(sce.pbmc,
features = "PF4",
x = "cluster", colour_by = "cluster"
)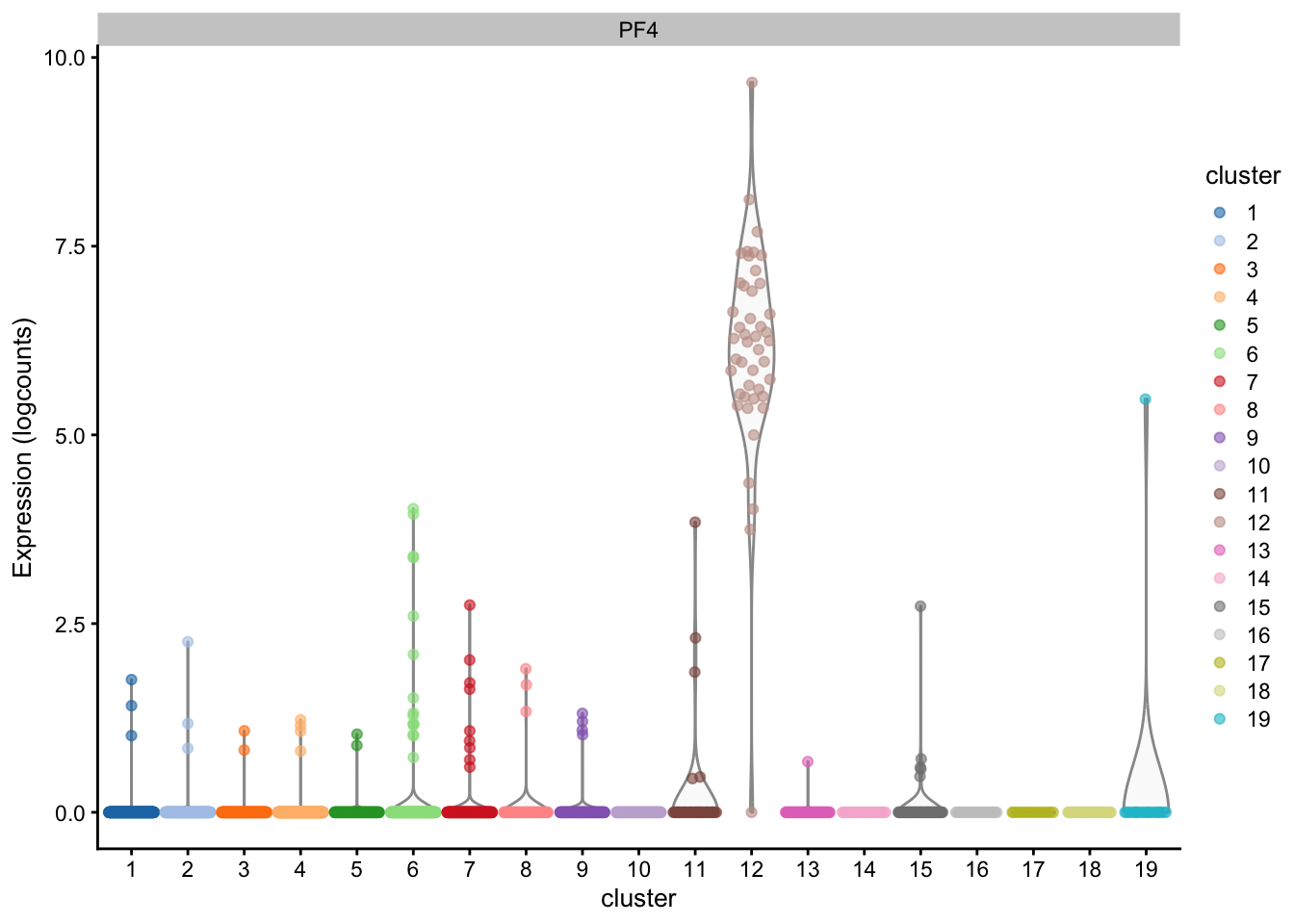
# El gen PF4 (platelet factor 4) solo se expresa en el cluster 9, sugiere que ese cluster son plateletas (trombocitos) células que forman coágulos sólidos y frenan hemorragias y cierran heridas# El gen CD3E está asociado con el clustering?
plotExpression(sce.pbmc,
features = "CD3E",
x = "cluster", colour_by = "cluster"
)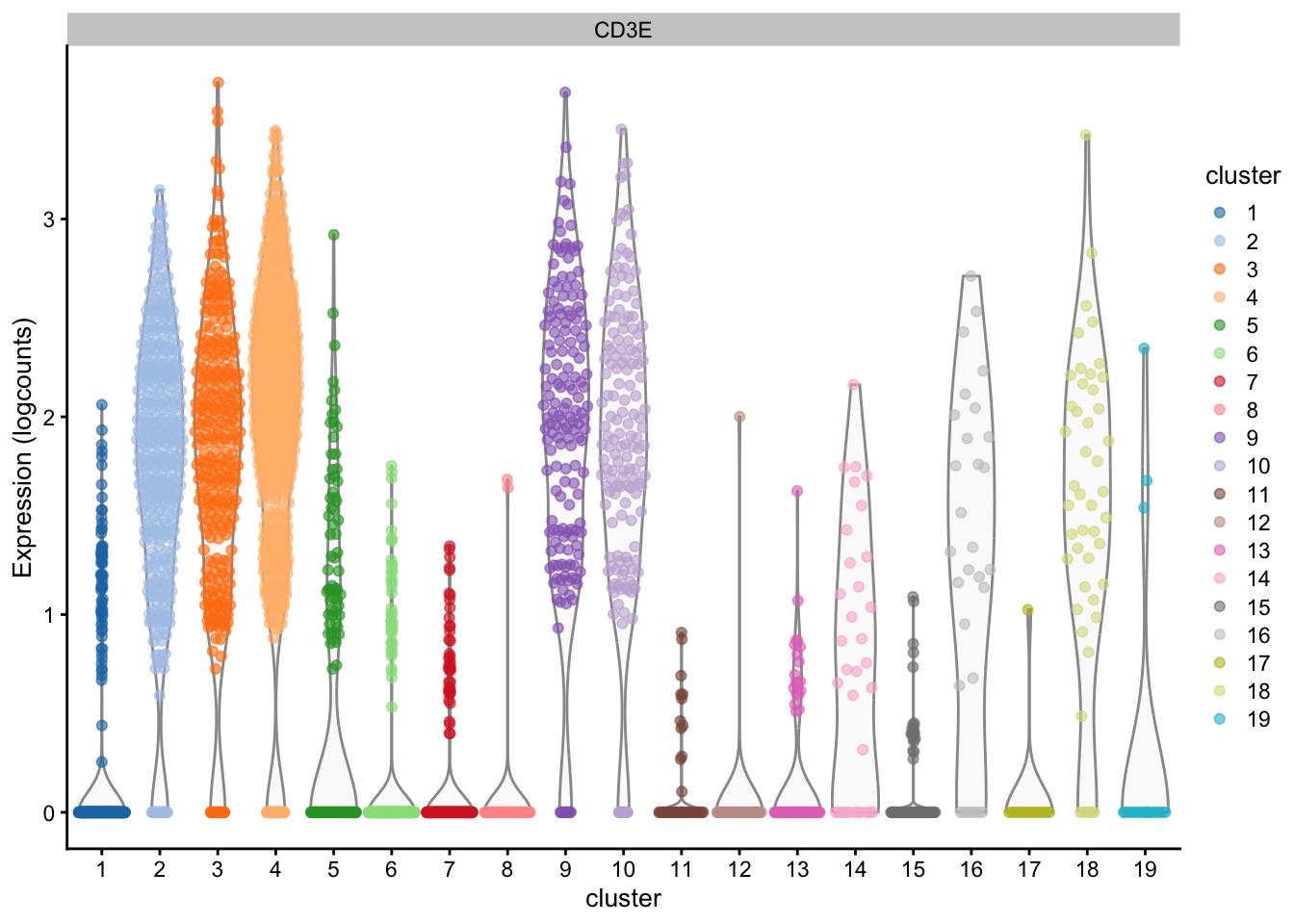
Ver una gráfica como una forma de encontrar los genes marcadores obviamente no nos sirve a gran escala
Necesitamos un método estadístico para identificar estos genes marcadores
👉 La prueba t de Welch parece una opción obvia para probar las diferencias en la expresión entre clústeres
7.5 Prueba t modificada de Welch pareada
➕ Rápidas y buenas propiedades estadísticas para un gran número de células (Soneson and Robinson, 2018) 6
➕ Las comparaciones pareadas proveen un log-fold change para indicar cuáles clústeres son distinguidos por cada gen
🤔 ¿Por qué no comparar cada clúster con el promedio de todas las otras células?
- Sensible a la composición poblacional, una subpoblación dominante sola que dirige la selección de los marcadores top para cualquier otro clúster
7.6 Ejemplo ilustrativo: CD3E como gen marcador en el dataset PBMC4k 10X
7.6.1 Pruebas pareadas
| comparison | logFC | Pval |
|---|---|---|
| 1 vs 2 | 1.50 | 1.7e-198 |
| 1 vs 3 | -0.08 | 0.11 |
| … | … | … |
| 1 vs 18 | 1.39 | 2.2e-9 |
| 2 vs 1 | -1.50 | 1.7e-198 |
K = 18 clústeres K!/(K-2)! = 306 comparaciones * La mitad de ellas son redundantes
7.6.2 Combinando comparaciones del gen CD3E para el clúster 1
“Me interesa saber si el gen CD3E está diferencialmente expresado entre el clúster 1 y ..”
- cualquier (any) otro clúster = P = 1.3 x 10-205 (Simes adjusted P-value)
- todos (all) los otros clústeres = P = 0.11 (Berger’s intersection-union test)
- algunos (some) de los otros clústeres = P = 2.0 x 10-44 (mediana u otro cuantil, Holm-adjusted P-values)
7.6.3 Extendiendo a todos los genes
# scran::pairwiseTTests() # Para realizar un prueba t-test Welch (pares)
# scran::combineMarkers() # Para realizar las compaaciones que involucran clusters, aquí está la info de que significa any, all y some- M = 33,694 genes
- 🤓 K x M = 10,310,364 pruebas
- Comparaciones involucrando clúster 1…
- Comparaciones involucrando clúster …
- Comparaciones involucrando clúster 18
7.7 Aplicación estándar
Para cada clúster, usar pruebas t de Welch para identificar los genes que están diferencialmente expresados entre éste y cualquier (any) otro clúster
# scran::findMarkers()
library(scran)
markers.pbmc <- findMarkers(sce.pbmc, groups=sce.pbmc$cluster,
test.type="t", pval.type="any")
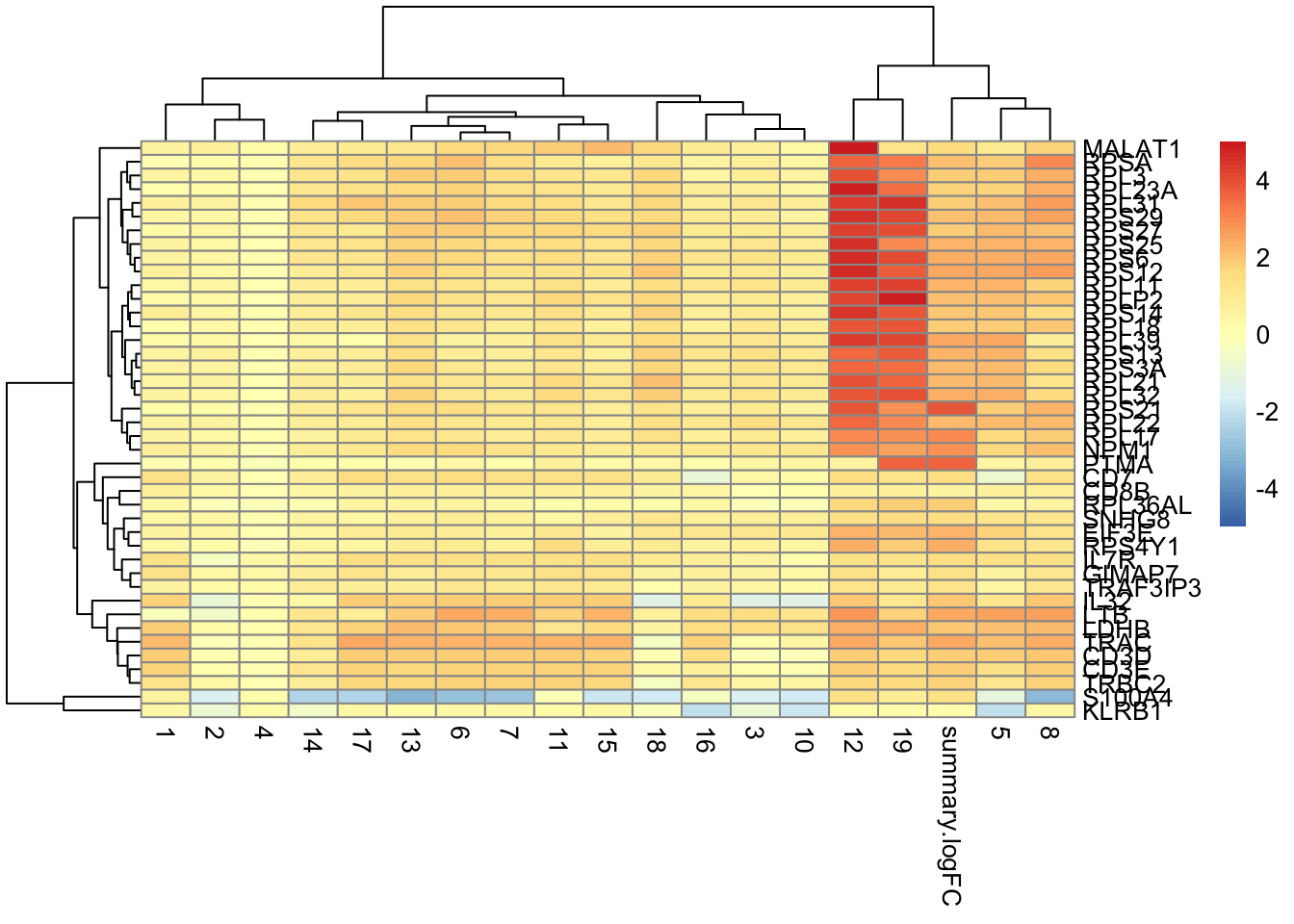
7.7.2 Con un heatmap
best.set <- interesting[interesting$Top <= 6,]
logFCs <- as.matrix(best.set[,-(1:3)])
colnames(logFCs) <- sub("logFC.", "", colnames(logFCs))
library(pheatmap)
pheatmap(logFCs, breaks=seq(-5, 5, length.out=101))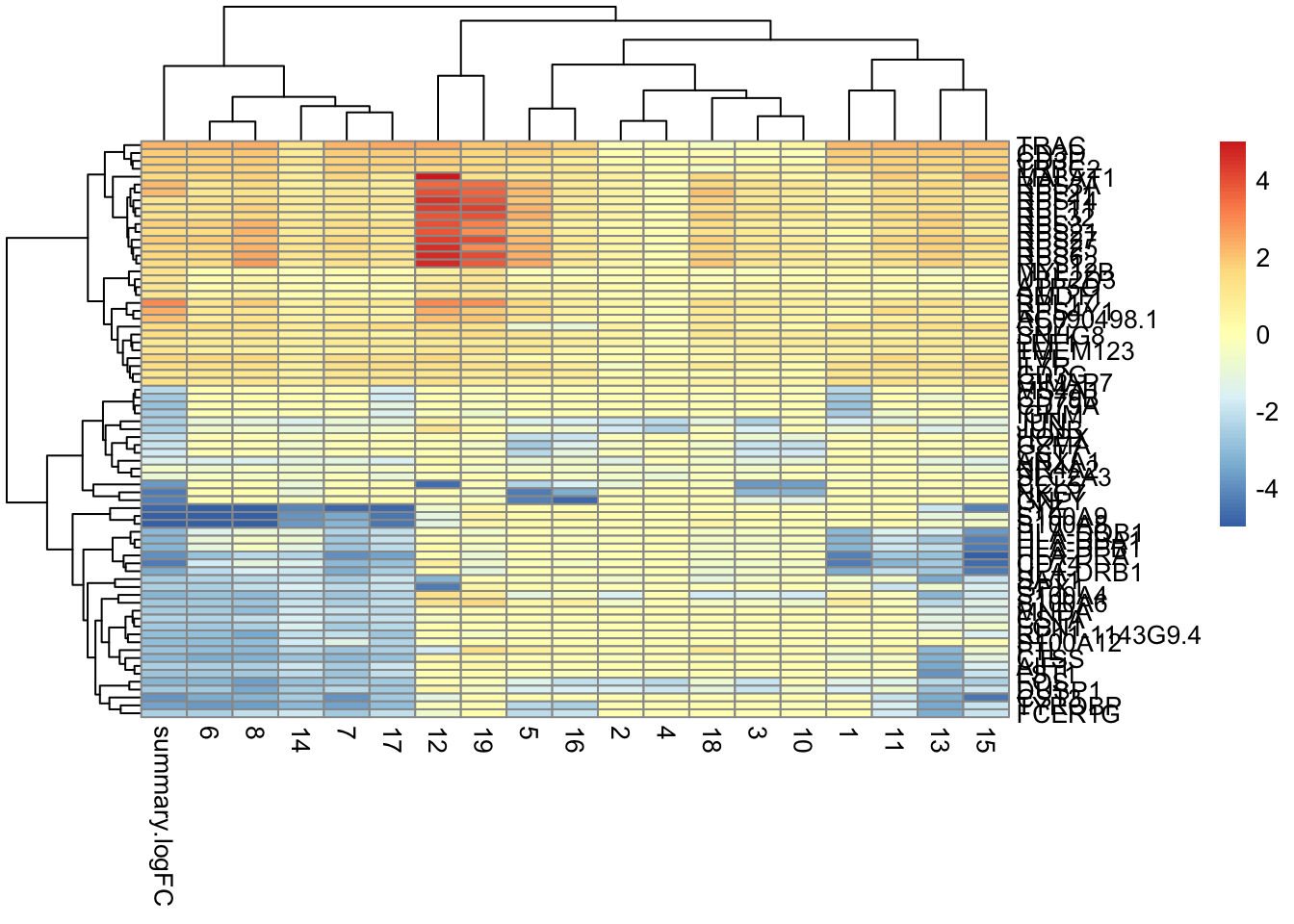
👉 Usamos el campo Top para identificar un conjunto de genes que distinguen al clúster 9 de cualquier otro clúster.
El conjunto de genes con Top <= X es la unión del top X genes (ordenados por p-value) de cada comparación de pares que involucran al cluster 9. Por ejemplo, el conjunto de todos los genes con valores Top 1 contienen el gen con el p-value mas pequeño de cada comparación. El conjunto de genes Top valor menor o igual a 10 contiene los 10 genes top de cada comparación. Se consolida en un ranking para cada clúster.
7.8 Usando el log-fold change
7.8.1 Sin especificar el lfc
Para cada clúster, usa pruebas t de Welch para identificar los genes que están sobreexpresados entre éste y cualquier otro clúster
7.8.2 Usando el lfc
Para cada clúster, usa pruebas t de Welch para identificar los genes que están sobreexpresados con un log-fold change (lfc) o al menos 1 entre éste y cualquier otro clúster
markers.pbmc.up2 <- findMarkers(sce.pbmc, groups=sce.pbmc$cluster,
test.type="t", direction="up", lfc=1, pval.type="any")
interesting.up2 <- markers.pbmc.up2[[chosen]]- 👉 La prueba t también nos permite especificar un log-fold change diferente de cero como la hipótesis nula
- 🤓 Es más riguroso que simplemente filtrar por log-fold change (TREAT) 7
7.8.3 Heatmap
best.set <- interesting.up2[interesting.up2$Top <= 5,]
logFCs <- as.matrix(best.set[,-(1:3)])
colnames(logFCs) <- sub("logFC.", "", colnames(logFCs))
pheatmap(logFCs, breaks=seq(-5, 5, length.out=101))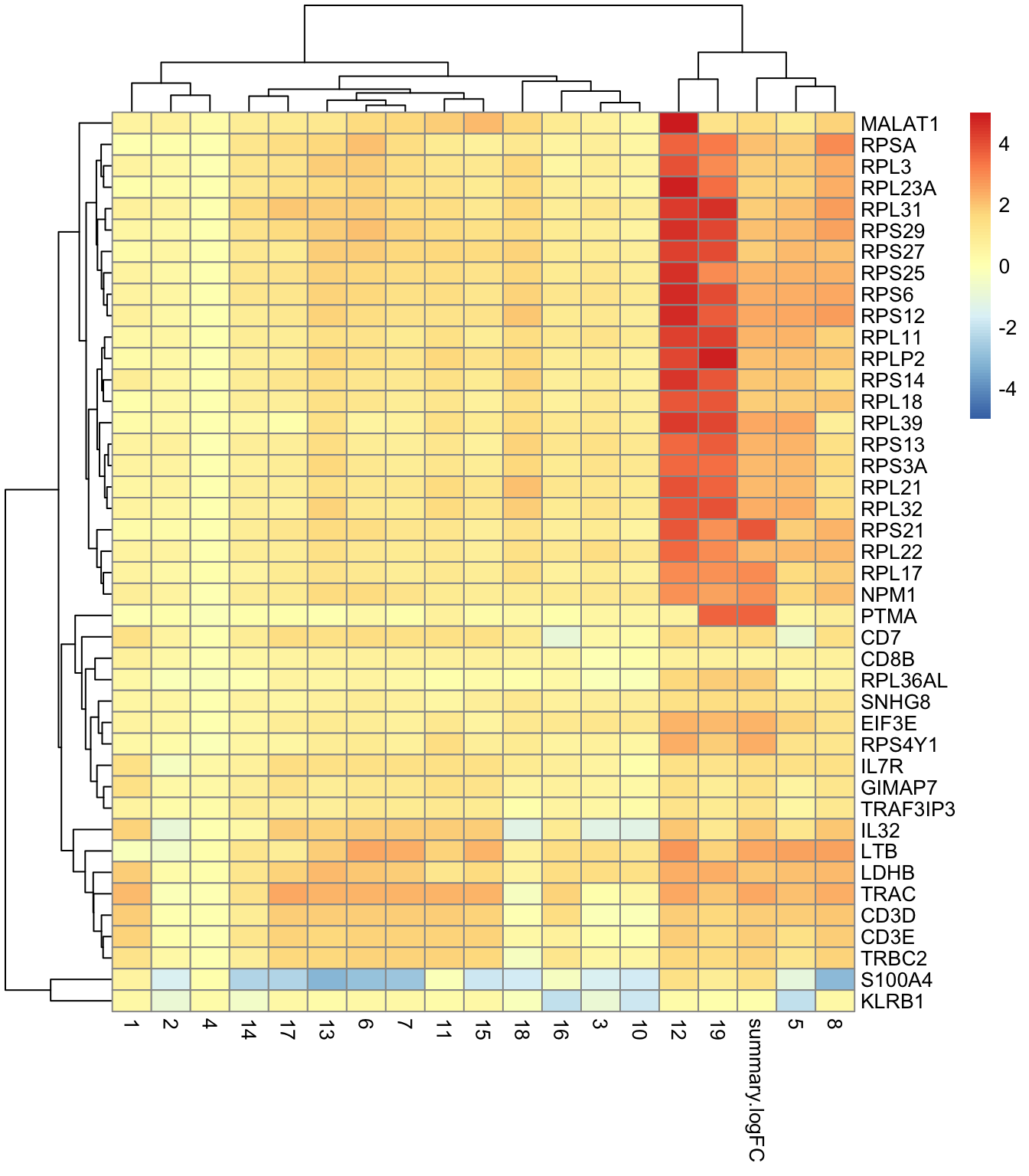
- 👉 Los promedios están más centrados en un conjunto de genes marcadores candidatos que están sobreexpresados en el clúster 9
- ⚠️ El incremento del rigor no se da sin costo
- ⚠️ Si el lfc es muy grande podría descartar genes útiles
- E.g., un gen sobreexpresado en una proporción pequeña de células en un clúster sigue siendo un marcador efectivo si el foco está en la especificidad más que en la sensibilidad
7.9 Encontrando marcadores específicos de clústeres
- 👉 Por defecto,
scran::findMarkers()dará un alto rango a genes que están DE en cualquier comparación pareada - 🤔 Quiero genes que son específicos de cada clúster
- 👉 Tú quieres genes que son DE en todas las comparaciones pareadas
Para cada clúster, usa pruebas t de Welch para identificar genes que están sobreexpresados entre éste y todos los otros clústeres
Considera todos los genes que están diferencialmente expresados en todas las comparaciones pareadas incluyendo al clúster de interés
markers.pbmc.up3 <- findMarkers(sce.pbmc, groups=sce.pbmc$cluster,
direction="up", pval.type="all")
interesting.up3 <- markers.pbmc.up3[[chosen]]🤓 Usa una prueba de unión-intersección para combinar los P-values que es el máximo P-value de todas las comparaciones pareadas.
Un gen solo logrará un bajo nivel de P-value combinado si está fuertemente DE en todas las comparaciones - cuando funciona es muy efectivo - te da un pequeño conjunto de marcadores candidatos.
Un gen que esté al mismo nivel que los demás clústeres no será detectado. No nos serviría para diferenciar en las poblaciones CD4 CD8.
7.9.1 Pros/cons de los genes marcadores específicos de los clústeres
| Poblacion | Expresion_CD4 | Expresion_CD8 |
|---|---|---|
| DN(CD4-/CD8-) | No | No |
| CD4+> | Si | No |
| CD8+> | No | Si |
| DP(CD4+/CD8+) | Si | Si |
7.9.2 findMarkers con pval.type some
Para cada clúster, usa pruebas t de Welch para identificar los genes que están sobreexpresados entre éste y algunos de los otros clústers.
markers.pbmc.up4 <- findMarkers(sce.pbmc, groups=sce.pbmc$cluster,
direction="up", pval.type="some")
interesting.up4 <- markers.pbmc.up4[[chosen]]- 👉 Si
pval.type="all"es muy estricto, entoncespval.type="any"es muy generoso - 🤓 Podemos entonces usar some. Aplica la corrección Holm-Bonferroni a los P-values y toma el mejor valor de en medio como el P-value combinado
Prueba la hipótesis nula de que al menos 50% de las comparaciones pareadas no tienen DE Se aplica un rango para obtener el conjunto de los mejores marcadores
- ⚠️ Perderás algunas garantías ofrecidas por los otros métodos
7.10 Pruebas alternas
7.10.1 Motivación
La prueba t no es la única forma de comparar dos grupos de mediciones
🤔 Quiero una prueba que pueda ser usada perfectamente para distinguir dos clústeres uno del otro
👉 Prueba de rangos Wilcoxon
🤔 Quiero identificar genes que son expresados más frecuentemente en un clúster que en otro
👉 Prueba Binomial
7.11 Prueba de rangos de Wilcoxon
Evalúa directamente la separación entre la distribución de la expresión de los diferentes clústeres
🤓 Es proporcional al área bajo la curva (AUC), que es la probabilidad de que una célula al azar de un clúster tenga mayor que expresión que una célula al azar de otro clúster
👉 AUCs de 1 o 0 indican que los dos clústeres tienen distribuciones de expresión separadas
🤓 También se conoce como prueba Wilcoxon-Mann-Whitney (WMW)
7.11.1 findMarkers para Wilcoxon
Para cada clúster, usa la prueba de rangos de Wilcoxon para identificar genes que están sobreexpresados entre éste y cualquier otro clúster
7.11.3 Resumen de la prueba de rangos de Wilcoxon
- ➕ Ofrece directamente la propiedad deseable de un gen marcador (i.e. que el gen distinga perfectamente entre dos clústeres)
- ➕ Es simétrico con respecto a las diferencias en el tamaño de los grupos comparados
- ➖ Es mucho más lento comparado con la prueba t (aunque esto en general no es un problema en la práctica)
7.12 Prueba binomial
Es una prueba que identifica los genes que difieren en la proporción de células que se expresan entre clústeres
Una definición mucho más estricta de genes marcadores
🤓 Convierte la expresión en una medida binaria de presencia/ausencia, por lo que toda la información cuantitativa es ignorada
Desde una perspectiva práctica, puede ser más fácil para validar
7.12.1 findMarkers para binomial
Para cada clúster, usa la prueba Binomial para identificar genes que están más frecuentemente expresados (sobreexpresados) en comparación con cualquier otro clúster
markers.pbmc.binom <- findMarkers(sce.pbmc,
groups=sce.pbmc$cluster, test.type="binom",
direction="up", pval.type="any")
interesting.binom <- markers.pbmc.binom[[chosen]]- 🤓 El efecto en el tamaño se reporta como el log-fold change en la proporción de las células que se expresan entre clústeres
- 👉 Log-fold changes grandes positivos, indican que el gen está más frecuentemente expresado en un clúster comparado con otro
7.13 Métodos de expresión diferencial personalizados
🤔 ¿Por qué no usar edgeR/DESeq2/limma-voom u otros métodos personalizados (e.g., MAST)?
👉 Claro que puedes! Checa OSCA
👉 Pero éstos son tal vez algo exagerados para identificar genes marcadores
🤓 Las células son nuestras “réplicas” para el propósito de identificar genes marcadores
🤓 edgeR/DESeq2/limma-voom hacen asunciones más fuertes acerca de los datos que es más probable que no se cumplan para células individuales en scRNA-seq
7.14 Problemas estadísticos
7.14.1 Invalidez de P-values
Todas las estrategias de DE para detectar genes marcadores entre clústeres son estadísticamente defectuosas de alguna manera
🤓 “Dragado de datos”: El análisis DE se realiza usando los mismos datos usados para obtener los clústeres
👉 Las pruebas para genes DE entre clústeres producirá inevitablemente algunos resultados significativos y así es como los clústeres serán definidos!
👉 Aún cuando los P-values son defectuosos, el efecto no es muy dañino para la detección de genes ya que los P-values solo son usados para los rangos
🤓 No se pueden usar P-values para definir “diferencias significativas” entre los clústeres con respecto a un umbral de la tasa de error
7.14.2 Naturaleza de la replicación
👉 Idealmente, validar algunos de los marcadores con una población de células independientes (y idealmente usando una técnica diferente, e.g., hibridación fluorescente in situ o qPCR)
7.14.3 Comentarios adicionales
- 👉 La estrategia de análisis DE es que los marcadores son definidos relativo a subpoblaciones en el mismo dataset
- 👉 Si un gen se expresa uniformemente a través de la población no servirá como un marcador
- e.g., los marcadores de las células T no serán detectados si solamente hay células T en los datos
- usualmente no es un problema, ya que tenemos idea de las células que se capturaron
- 👉 Existen métodos de machine learning para hacer la identificación de los genes marcadores, pero la humilde prueba t sigue siendo muy buena
7.14.4 Resumen y recomendaciones
- 👉 Crea múltiples listas de genes marcadores con diferentes niveles de rigor
- 👉 La forma más simple de interpretar los genes marcadores es que son los sobreexpresados de “forma única”, o son “genes específicos de clústeres”, especialmente si queremos imponer un log-fold change mínimo
- 👉 Puedes requerir hacer una identificación de genes marcadores más enfocada, e.g., subset de los datos de solo 2 clústeres de interés y entonces correr
scran::findMarkers()
7.14.5 Nota sobre la nueva versión en OSCA
## List of length 19
## names(19): 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19Compara cada par de clusters, y calcula scores cuantificando las diferencias en las distribuciones de la expresión entre clusters
## [1] "self.average" "other.average" "self.detected" "other.detected"
## [5] "mean.logFC.cohen" "min.logFC.cohen" "median.logFC.cohen" "max.logFC.cohen"
## [9] "rank.logFC.cohen" "mean.AUC" "min.AUC" "median.AUC"
## [13] "max.AUC" "rank.AUC" "mean.logFC.detected" "min.logFC.detected"
## [17] "median.logFC.detected" "max.logFC.detected" "rank.logFC.detected"## DataFrame with 6 rows and 4 columns
## self.average other.average self.detected other.detected
## <numeric> <numeric> <numeric> <numeric>
## CD79A 2.97173 0.153167 0.992620 0.124460
## CD79B 2.82857 0.315276 0.977860 0.248526
## CD37 3.33505 1.417883 1.000000 0.785371
## MS4A1 2.29606 0.147344 0.952030 0.103018
## CD74 5.93908 2.835638 1.000000 0.887106
## IGHM 2.63948 0.230242 0.896679 0.166787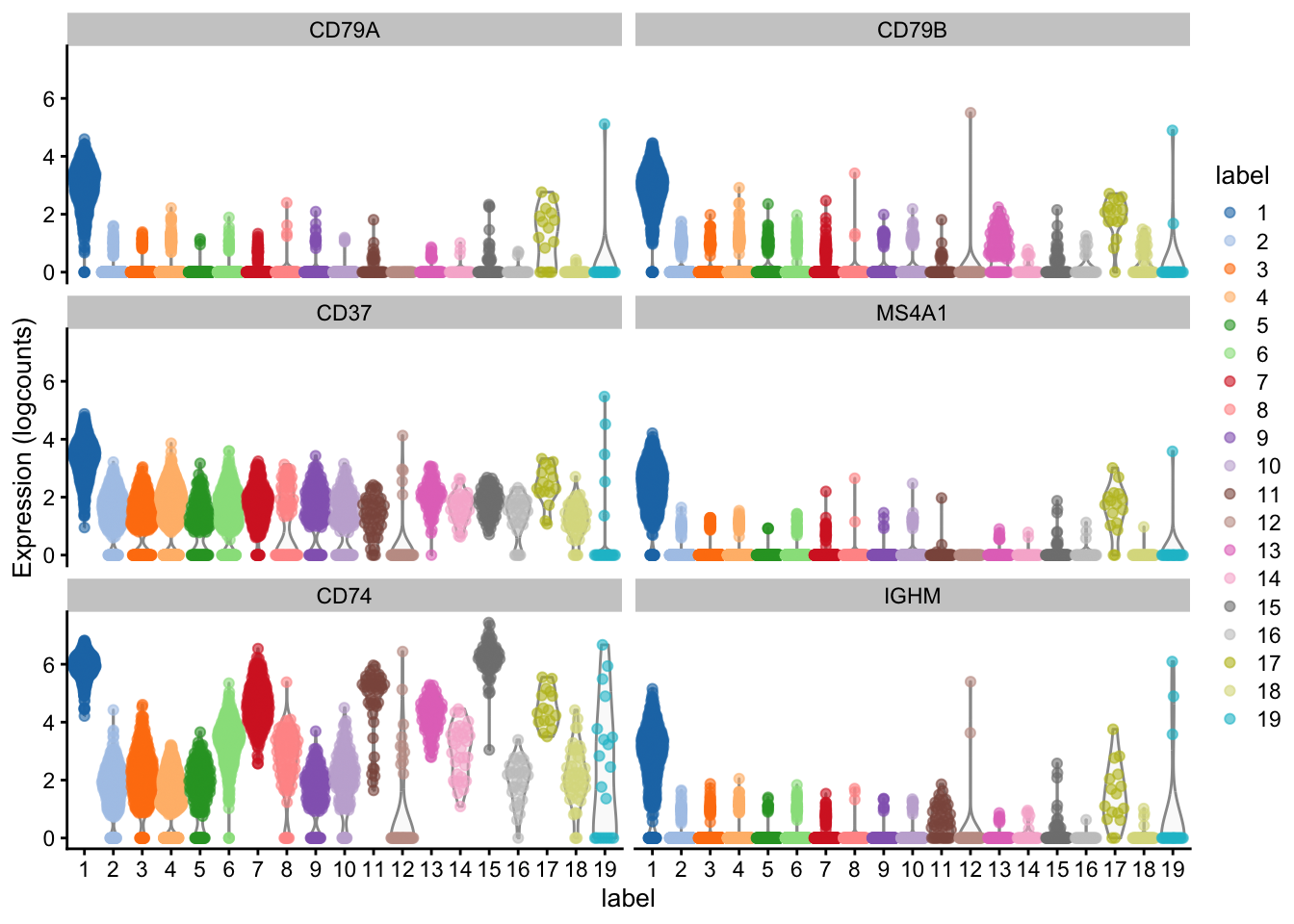
7.15 Detalles de la sesión de R
## ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
## setting value
## version R version 4.3.1 (2023-06-16)
## os macOS Ventura 13.4.1
## system aarch64, darwin20
## ui RStudio
## language (EN)
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz America/New_York
## date 2023-08-09
## rstudio 2023.06.0+421 Mountain Hydrangea (desktop)
## pandoc 3.1.1 @ /Applications/RStudio.app/Contents/Resources/app/quarto/bin/tools/ (via rmarkdown)
##
## ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
## package * version date (UTC) lib source
## abind 1.4-5 2016-07-21 [1] CRAN (R 4.3.0)
## AnnotationDbi * 1.62.2 2023-07-02 [1] Bioconductor
## AnnotationFilter * 1.24.0 2023-05-08 [1] Bioconductor
## AnnotationHub * 3.8.0 2023-05-08 [1] Bioconductor
## beachmat 2.16.0 2023-05-08 [1] Bioconductor
## beeswarm 0.4.0 2021-06-01 [1] CRAN (R 4.3.0)
## Biobase * 2.60.0 2023-05-08 [1] Bioconductor
## BiocFileCache * 2.8.0 2023-05-08 [1] Bioconductor
## BiocGenerics * 0.46.0 2023-06-04 [1] Bioconductor
## BiocIO 1.10.0 2023-05-08 [1] Bioconductor
## BiocManager 1.30.21.1 2023-07-18 [1] CRAN (R 4.3.0)
## BiocNeighbors 1.18.0 2023-05-08 [1] Bioconductor
## BiocParallel 1.34.2 2023-05-28 [1] Bioconductor
## BiocSingular 1.16.0 2023-05-08 [1] Bioconductor
## BiocVersion 3.17.1 2022-12-20 [1] Bioconductor
## biomaRt 2.56.1 2023-06-11 [1] Bioconductor
## Biostrings 2.68.1 2023-05-21 [1] Bioconductor
## bit 4.0.5 2022-11-15 [1] CRAN (R 4.3.0)
## bit64 4.0.5 2020-08-30 [1] CRAN (R 4.3.0)
## bitops 1.0-7 2021-04-24 [1] CRAN (R 4.3.0)
## blob 1.2.4 2023-03-17 [1] CRAN (R 4.3.0)
## bluster * 1.10.0 2023-05-08 [1] Bioconductor
## bookdown 0.34 2023-05-09 [1] CRAN (R 4.3.0)
## bslib 0.5.0 2023-06-09 [1] CRAN (R 4.3.0)
## cachem 1.0.8 2023-05-01 [1] CRAN (R 4.3.0)
## cli 3.6.1 2023-03-23 [1] CRAN (R 4.3.0)
## cluster 2.1.4 2022-08-22 [1] CRAN (R 4.3.1)
## codetools 0.2-19 2023-02-01 [1] CRAN (R 4.3.1)
## colorspace 2.1-0 2023-01-23 [1] CRAN (R 4.3.0)
## cowplot 1.1.1 2020-12-30 [1] CRAN (R 4.3.0)
## crayon 1.5.2 2022-09-29 [1] CRAN (R 4.3.0)
## curl 5.0.1 2023-06-07 [1] CRAN (R 4.3.0)
## DBI 1.1.3 2022-06-18 [1] CRAN (R 4.3.0)
## dbplyr * 2.3.3 2023-07-07 [1] CRAN (R 4.3.0)
## DelayedArray 0.26.7 2023-07-28 [1] Bioconductor
## DelayedMatrixStats 1.22.1 2023-06-09 [1] Bioconductor
## digest 0.6.33 2023-07-07 [1] CRAN (R 4.3.0)
## dplyr * 1.1.2 2023-04-20 [1] CRAN (R 4.3.0)
## dqrng 0.3.0 2021-05-01 [1] CRAN (R 4.3.0)
## DropletUtils * 1.20.0 2023-05-08 [1] Bioconductor
## edgeR 3.42.4 2023-06-04 [1] Bioconductor
## ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.3.0)
## EnsDb.Hsapiens.v86 * 2.99.0 2023-07-29 [1] Bioconductor
## ensembldb * 2.24.0 2023-05-08 [1] Bioconductor
## evaluate 0.21 2023-05-05 [1] CRAN (R 4.3.0)
## ExperimentHub 2.8.1 2023-07-16 [1] Bioconductor
## fansi 1.0.4 2023-01-22 [1] CRAN (R 4.3.0)
## farver 2.1.1 2022-07-06 [1] CRAN (R 4.3.0)
## fastmap 1.1.1 2023-02-24 [1] CRAN (R 4.3.0)
## filelock 1.0.2 2018-10-05 [1] CRAN (R 4.3.0)
## FNN 1.1.3.2 2023-03-20 [1] CRAN (R 4.3.0)
## generics 0.1.3 2022-07-05 [1] CRAN (R 4.3.0)
## GenomeInfoDb * 1.36.1 2023-07-02 [1] Bioconductor
## GenomeInfoDbData 1.2.10 2023-06-08 [1] Bioconductor
## GenomicAlignments 1.36.0 2023-05-08 [1] Bioconductor
## GenomicFeatures * 1.52.1 2023-07-02 [1] Bioconductor
## GenomicRanges * 1.52.0 2023-05-08 [1] Bioconductor
## ggbeeswarm 0.7.2 2023-04-29 [1] CRAN (R 4.3.0)
## ggplot2 * 3.4.2 2023-04-03 [1] CRAN (R 4.3.0)
## ggrepel * 0.9.3 2023-02-03 [1] CRAN (R 4.3.0)
## glue 1.6.2 2022-02-24 [1] CRAN (R 4.3.0)
## gridExtra 2.3 2017-09-09 [1] CRAN (R 4.3.0)
## gtable 0.3.3 2023-03-21 [1] CRAN (R 4.3.0)
## HDF5Array 1.28.1 2023-05-08 [1] Bioconductor
## here 1.0.1 2020-12-13 [1] CRAN (R 4.3.0)
## highr 0.10 2022-12-22 [1] CRAN (R 4.3.0)
## hms 1.1.3 2023-03-21 [1] CRAN (R 4.3.0)
## htmltools 0.5.5 2023-03-23 [1] CRAN (R 4.3.0)
## httpuv 1.6.11 2023-05-11 [1] CRAN (R 4.3.0)
## httr 1.4.6 2023-05-08 [1] CRAN (R 4.3.0)
## igraph 1.5.0.1 2023-07-23 [1] CRAN (R 4.3.0)
## interactiveDisplayBase 1.38.0 2023-05-08 [1] Bioconductor
## IRanges * 2.34.1 2023-07-02 [1] Bioconductor
## irlba 2.3.5.1 2022-10-03 [1] CRAN (R 4.3.0)
## jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.3.0)
## jsonlite 1.8.7 2023-06-29 [1] CRAN (R 4.3.0)
## kableExtra * 1.3.4 2021-02-20 [1] CRAN (R 4.3.0)
## KEGGREST 1.40.0 2023-05-08 [1] Bioconductor
## knitr 1.43 2023-05-25 [1] CRAN (R 4.3.0)
## labeling 0.4.2 2020-10-20 [1] CRAN (R 4.3.0)
## later 1.3.1 2023-05-02 [1] CRAN (R 4.3.0)
## lattice 0.21-8 2023-04-05 [1] CRAN (R 4.3.1)
## lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.3.0)
## lifecycle 1.0.3 2022-10-07 [1] CRAN (R 4.3.0)
## limma 3.56.2 2023-06-04 [1] Bioconductor
## locfit 1.5-9.8 2023-06-11 [1] CRAN (R 4.3.0)
## magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.3.0)
## Matrix * 1.6-0 2023-07-08 [1] CRAN (R 4.3.0)
## MatrixGenerics * 1.12.3 2023-07-30 [1] Bioconductor
## matrixStats * 1.0.0 2023-06-02 [1] CRAN (R 4.3.0)
## memoise 2.0.1 2021-11-26 [1] CRAN (R 4.3.0)
## metapod 1.8.0 2023-04-25 [1] Bioconductor
## mime 0.12 2021-09-28 [1] CRAN (R 4.3.0)
## munsell 0.5.0 2018-06-12 [1] CRAN (R 4.3.0)
## patchwork * 1.1.2 2022-08-19 [1] CRAN (R 4.3.0)
## PCAtools * 2.12.0 2023-05-08 [1] Bioconductor
## pheatmap * 1.0.12 2019-01-04 [1] CRAN (R 4.3.0)
## pillar 1.9.0 2023-03-22 [1] CRAN (R 4.3.0)
## pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.3.0)
## plyr 1.8.8 2022-11-11 [1] CRAN (R 4.3.0)
## png 0.1-8 2022-11-29 [1] CRAN (R 4.3.0)
## prettyunits 1.1.1 2020-01-24 [1] CRAN (R 4.3.0)
## progress 1.2.2 2019-05-16 [1] CRAN (R 4.3.0)
## promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.3.0)
## ProtGenerics 1.32.0 2023-05-08 [1] Bioconductor
## purrr 1.0.1 2023-01-10 [1] CRAN (R 4.3.0)
## R.methodsS3 1.8.2 2022-06-13 [1] CRAN (R 4.3.0)
## R.oo 1.25.0 2022-06-12 [1] CRAN (R 4.3.0)
## R.utils 2.12.2 2022-11-11 [1] CRAN (R 4.3.0)
## R6 2.5.1 2021-08-19 [1] CRAN (R 4.3.0)
## rappdirs 0.3.3 2021-01-31 [1] CRAN (R 4.3.0)
## RColorBrewer 1.1-3 2022-04-03 [1] CRAN (R 4.3.0)
## Rcpp 1.0.11 2023-07-06 [1] CRAN (R 4.3.0)
## RCurl 1.98-1.12 2023-03-27 [1] CRAN (R 4.3.0)
## reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.3.0)
## restfulr 0.0.15 2022-06-16 [1] CRAN (R 4.3.0)
## rhdf5 2.44.0 2023-05-08 [1] Bioconductor
## rhdf5filters 1.12.1 2023-05-08 [1] Bioconductor
## Rhdf5lib 1.22.0 2023-05-08 [1] Bioconductor
## rjson 0.2.21 2022-01-09 [1] CRAN (R 4.3.0)
## rlang 1.1.1 2023-04-28 [1] CRAN (R 4.3.0)
## rmarkdown 2.23 2023-07-01 [1] CRAN (R 4.3.0)
## rprojroot 2.0.3 2022-04-02 [1] CRAN (R 4.3.0)
## Rsamtools 2.16.0 2023-06-04 [1] Bioconductor
## RSQLite 2.3.1 2023-04-03 [1] CRAN (R 4.3.0)
## rstudioapi 0.15.0 2023-07-07 [1] CRAN (R 4.3.0)
## rsvd 1.0.5 2021-04-16 [1] CRAN (R 4.3.0)
## rtracklayer 1.60.0 2023-05-08 [1] Bioconductor
## Rtsne 0.16 2022-04-17 [1] CRAN (R 4.3.0)
## rvest 1.0.3 2022-08-19 [1] CRAN (R 4.3.0)
## S4Arrays 1.0.5 2023-07-24 [1] Bioconductor
## S4Vectors * 0.38.1 2023-05-08 [1] Bioconductor
## sass 0.4.7 2023-07-15 [1] CRAN (R 4.3.0)
## ScaledMatrix 1.8.1 2023-05-08 [1] Bioconductor
## scales 1.2.1 2022-08-20 [1] CRAN (R 4.3.0)
## scater * 1.28.0 2023-04-25 [1] Bioconductor
## scran * 1.28.2 2023-07-23 [1] Bioconductor
## scRNAseq * 2.14.0 2023-04-27 [1] Bioconductor
## scuttle * 1.9.4 2023-01-23 [1] Bioconductor
## sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.3.0)
## shiny 1.7.4.1 2023-07-06 [1] CRAN (R 4.3.0)
## SingleCellExperiment * 1.22.0 2023-05-08 [1] Bioconductor
## sparseMatrixStats 1.12.2 2023-07-02 [1] Bioconductor
## statmod 1.5.0 2023-01-06 [1] CRAN (R 4.3.0)
## stringi 1.7.12 2023-01-11 [1] CRAN (R 4.3.0)
## stringr 1.5.0 2022-12-02 [1] CRAN (R 4.3.0)
## SummarizedExperiment * 1.30.2 2023-06-06 [1] Bioconductor
## svglite 2.1.1 2023-01-10 [1] CRAN (R 4.3.0)
## systemfonts 1.0.4 2022-02-11 [1] CRAN (R 4.3.0)
## tibble 3.2.1 2023-03-20 [1] CRAN (R 4.3.0)
## tidyselect 1.2.0 2022-10-10 [1] CRAN (R 4.3.0)
## utf8 1.2.3 2023-01-31 [1] CRAN (R 4.3.0)
## uwot 0.1.16 2023-06-29 [1] CRAN (R 4.3.0)
## vctrs 0.6.3 2023-06-14 [1] CRAN (R 4.3.0)
## vipor 0.4.5 2017-03-22 [1] CRAN (R 4.3.0)
## viridis 0.6.4 2023-07-22 [1] CRAN (R 4.3.0)
## viridisLite 0.4.2 2023-05-02 [1] CRAN (R 4.3.0)
## webshot 0.5.5 2023-06-26 [1] CRAN (R 4.3.0)
## withr 2.5.0 2022-03-03 [1] CRAN (R 4.3.0)
## xfun 0.39 2023-04-20 [1] CRAN (R 4.3.0)
## XML 3.99-0.14 2023-03-19 [1] CRAN (R 4.3.0)
## xml2 1.3.5 2023-07-06 [1] CRAN (R 4.3.0)
## xtable 1.8-4 2019-04-21 [1] CRAN (R 4.3.0)
## XVector 0.40.0 2023-05-08 [1] Bioconductor
## yaml 2.3.7 2023-01-23 [1] CRAN (R 4.3.0)
## zlibbioc 1.46.0 2023-05-08 [1] Bioconductor
##
## [1] /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/library
##
## ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────Patrocinadores
La presentación fue hecha con el paquete de R xaringan y configurada con xaringanthemer.
Este curso está basado en el libro Orchestrating Single Cell Analysis with Bioconductor de Aaron Lun, Robert Amezquita, Stephanie Hicks y Raphael Gottardo, además del curso de scRNA-seq para WEHI creado por Peter Hickey.
Puedes encontrar los archivos para este taller en comunidadbioinfo/cdsb2023.
Descarga los materiales con usethis::use_course('comunidadbioinfo/cdsb2023') o revísalos en línea vía comunidadbioinfo.github.io/cdsb2023.
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).↩︎
Soneson C, Robinson MD. Bias, robustness and scalability in single-cell differential expression analysis. Nat Methods. 2018 Apr;15(4):255-261. doi: 10.1038/nmeth.4612. Epub 2018 Feb 26. PMID: 29481549.↩︎
McCarthy DJ, Smyth GK. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009 Mar 15;25(6):765-71. doi: 10.1093/bioinformatics/btp053. Epub 2009 Jan 28. PMID: 19176553; PMCID: PMC2654802.↩︎