5 Reducción de dimensiones
Instructora: Laura Gómez-Romero
5.1 Diapositivas de Peter Hickey
Contenido adaptado de: aquí
5.2 Motivación
El siguiente paso en el análisis de scRNA-seq usualmente consiste en identificar grupos de células “similares”.
Por ejemplo: un análisis de clustering busca identificar células con un perfil transcriptómico similar al calcular distancias entre ellas.
Si tuviéramos un dataset con dos genes podríamos hacer una gráfica de dos dimensiones para identificar clusters de células.
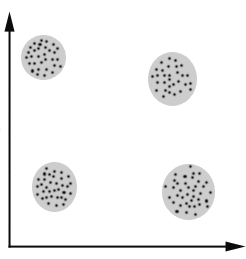
Pero… tenemos decenas de miles de genes : Reducción de dimensionalidad
5.3 Reducción de dimensionalidad
Es posible porque la expresión de diferentes genes estaría correlacionada si estos genes son afectados por el mismo proceso biológico.
Por lo tanto, no necesitamos almacenar información independiente para genes individuales. Podemos comprimir múltiples “features” (genes) en una única dimensión.
Ventajas:
- Reduce trabajo computacional en análisis posteriores.
- Reduce el ruido al “promediar” mútiples genes obteniendo una representación más precisa de los patrones en los datos.
- Permite una graficación efectiva en dos dimensiones.
5.4 Dataset ilustrativo: Zeisel
library(scRNAseq)
sce.zeisel <- ZeiselBrainData(ensembl = TRUE)
# Estos datos contienen tipos celulares previamente anotados
table(sce.zeisel$level1class)##
## astrocytes_ependymal endothelial-mural interneurons microglia oligodendrocytes
## 224 235 290 98 820
## pyramidal CA1 pyramidal SS
## 939 399Estudio de tipos celulares en el cerebro de ratón (oligodendrocitos, microglia, neuronas, etc) procesados con el sistema STRT-seq (similar a CEL-Seq)
Descripción aquí
Zeisel, A. et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138-1142 (2015)
# Quality control
# Descartar celulas con alto contenido mitocondrial o con alto porcentaje de spike-ins
library(scater)
is.mito <- which(rowData(sce.zeisel)$featureType == "mito")
stats <- perCellQCMetrics(sce.zeisel,
subsets = list(Mt = is.mito)
)
qc <- quickPerCellQC(stats,
percent_subsets = c("altexps_ERCC_percent", "subsets_Mt_percent")
)
sce.zeisel <- sce.zeisel[, !qc$discard]# normalization
# encontramos unos clusters rápidos para las células y usamos esa información para calcular los factores de tamaño
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.zeisel)
sce.zeisel <- computeSumFactors(sce.zeisel,
cluster = clusters
)
sce.zeisel <- logNormCounts(sce.zeisel)# variance-modelling
dec.zeisel <- modelGeneVarWithSpikes(
sce.zeisel,
"ERCC"
)
top.zeisel <- getTopHVGs(dec.zeisel, n = 2000)- ¿Cómo se está modelando la relación media varianza?
- ¿Cómo se están obteniendo los HGVs?
5.5 Dataset ilustrativo: 10x PBMC4k no filtradas
library(BiocFileCache)
bfc <- BiocFileCache()
raw.path <- bfcrpath(bfc, file.path(
"http://cf.10xgenomics.com/samples",
"cell-exp/2.1.0/pbmc4k/pbmc4k_raw_gene_bc_matrices.tar.gz"
))
untar(raw.path, exdir = file.path(tempdir(), "pbmc4k"))library(DropletUtils)
library(Matrix)
fname <- file.path(tempdir(), "pbmc4k/raw_gene_bc_matrices/GRCh38")
sce.pbmc <- read10xCounts(fname, col.names = TRUE)Dataset “Células mononucleares humanas de sangre periférica” de 10X Genomics
Descripción aquí
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017)
# gene-annotation
library(scater)
rownames(sce.pbmc) <- uniquifyFeatureNames(
rowData(sce.pbmc)$ID, rowData(sce.pbmc)$Symbol
)
library(EnsDb.Hsapiens.v86)
location <- mapIds(EnsDb.Hsapiens.v86,
keys = rowData(sce.pbmc)$ID,
column = "SEQNAME", keytype = "GENEID"
)
# cell-detection
set.seed(100)
e.out <- emptyDrops(counts(sce.pbmc))
sce.pbmc <- sce.pbmc[, which(e.out$FDR <= 0.001)]# quality-control
stats <- perCellQCMetrics(sce.pbmc,
subsets = list(Mito = which(location == "MT"))
)
high.mito <- isOutlier(stats$subsets_Mito_percent,
type = "higher"
)
sce.pbmc <- sce.pbmc[, !high.mito]
# normalization
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.pbmc)
sce.pbmc <- computeSumFactors(sce.pbmc, cluster = clusters)
sce.pbmc <- logNormCounts(sce.pbmc)# variance modelling
set.seed(1001)
dec.pbmc <- modelGeneVarByPoisson(sce.pbmc)
top.pbmc <- getTopHVGs(dec.pbmc, prop = 0.1)- ¿Cómo se está modelando la relación media varianza?
- ¿Cómo se están obteniendo los HGVs?
5.6 Análisis de Componentes Principales
PCA es el arma principal de la reducción de dimensionalidad.
PCA descubre las combinaciones (lineales) de “features” que capturan la cantidad más grande de variación
En un PCA, la primer combinación lineal (componente principal) se elige tal que permite capturar la mayor varianza a través de las células. El siguiente PC se elige tal que es “ortogonal” al primero y captura la cantidad más grande de la variación restante, y así sucesivamente…
5.6.1 PCA aplicado a datos de scRNA-seq
Podemos realizar reducción de dimensionalidad al aplicar PCA en la matriz de cuentas transformadas (log-counts matrix) y restringiendo los análisis posteriores a los primeros PCs (top PCs).
- Esto puede reducir nuestro dataset de 20,000 dimensiones a, digamos, 10, sin perder demasiada información.
- La técnica de PCA tiene muchas propiedades teóricas bien estudiadas.
- Hay varias formas rápidas de realizar PCA en datasets grandes.
5.6.2 Suposiciones de PCA aplicadas a los datos de scRNA-seq
- Los procesos biológicos afectan múltiples genes en una manera coordinada.
- Los primeros PCs probablemente representan la estructura biológica dado que más variación puede ser capturada considerando el comportamiento correlacionado de muchos genes.
- Se espera que el ruido técnico azaroso afecte cada gen independientemente.
Consideración: Los primeros PCs capturarón “batch effects” (efectos de lote) que afectan muchos genes en una manera coordinada
## Aplicar PCA en la matriz de cuentas transformadas (log-counts matrix)
library(scran)
library(scater)
set.seed(100)
sce.zeisel <- runPCA(sce.zeisel,
subset_row = top.zeisel
)¿Estamos corriendo el análisis sobre todos los genes de nuestro dataset?
Por default, runPCA() usa un método rápido aproximado que realiza simulaciones, por lo tanto, es necesario ‘configurar la semilla’ para obtener resultados reproducibles.
5.6.3 Eligiendo el número de PCs
Esta elección en análoga a la elección del número de highly variable genes (HGV). Elegir más PCs evitará descartar señal biológica a expensas de retener más ruido
- Es común seleccionar un número de PCs “razonable” pero arbitrario (10-50), continuar con el análisis y regresar para checar la robustez de los resultados en cierto rango de valores.
Ahora exploraremos algunas estrategias guiadas por los datos (data-driven) para hacer esta selección.
5.6.3.1 Usando el punto del codo
library(PCAtools)
percent.var <- attr(reducedDim(sce.zeisel), "percentVar")
chosen.elbow <- PCAtools::findElbowPoint(percent.var)
plot(percent.var, xlab = "PC", ylab = "Variance explained (%)")
abline(v = chosen.elbow, col = "red")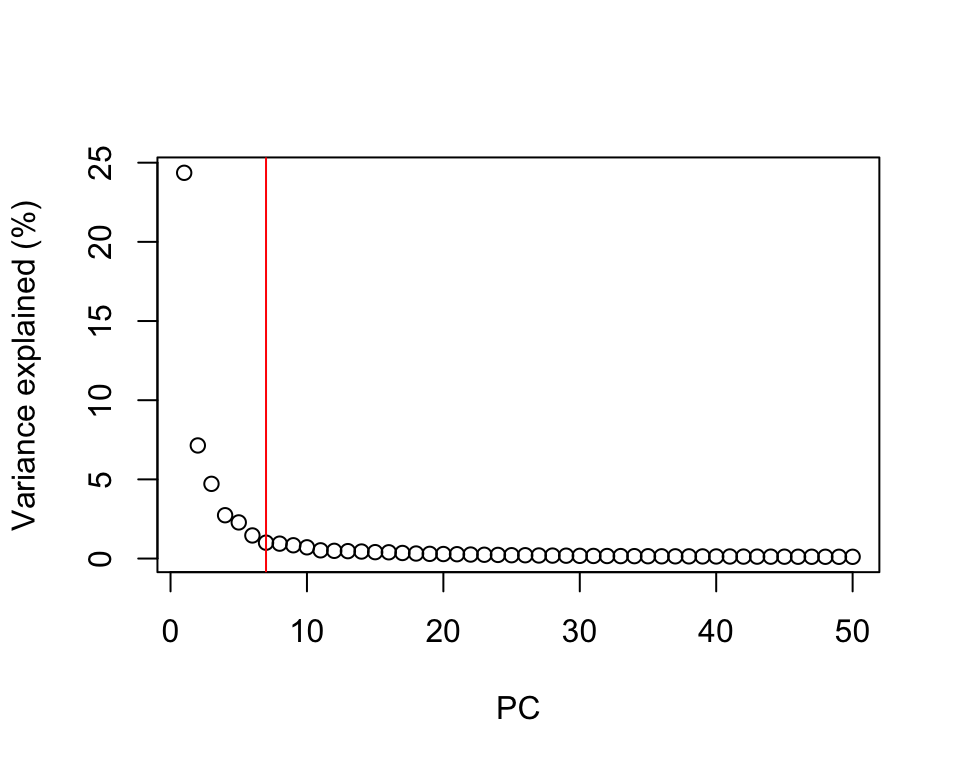
Una heurística simple es elegir el número de PCs basado en el porcentaje de varianza explicado por PCs sucesivos.
5.6.3.2 Basados en la estructura de la población
Esta es una aproximación heurística más sofisticada que usa el número de clusters como un proxy del número de subpoblaciones.
choices <- getClusteredPCs(reducedDim(sce.zeisel))
chosen.clusters <- metadata(choices)$chosen
plot(choices$n.pcs, choices$n.clusters,
xlab = "Number of PCs", ylab = "Number of clusters"
)
abline(a = 1, b = 1, col = "red")
abline(v = chosen.clusters, col = "grey80", lty = 2)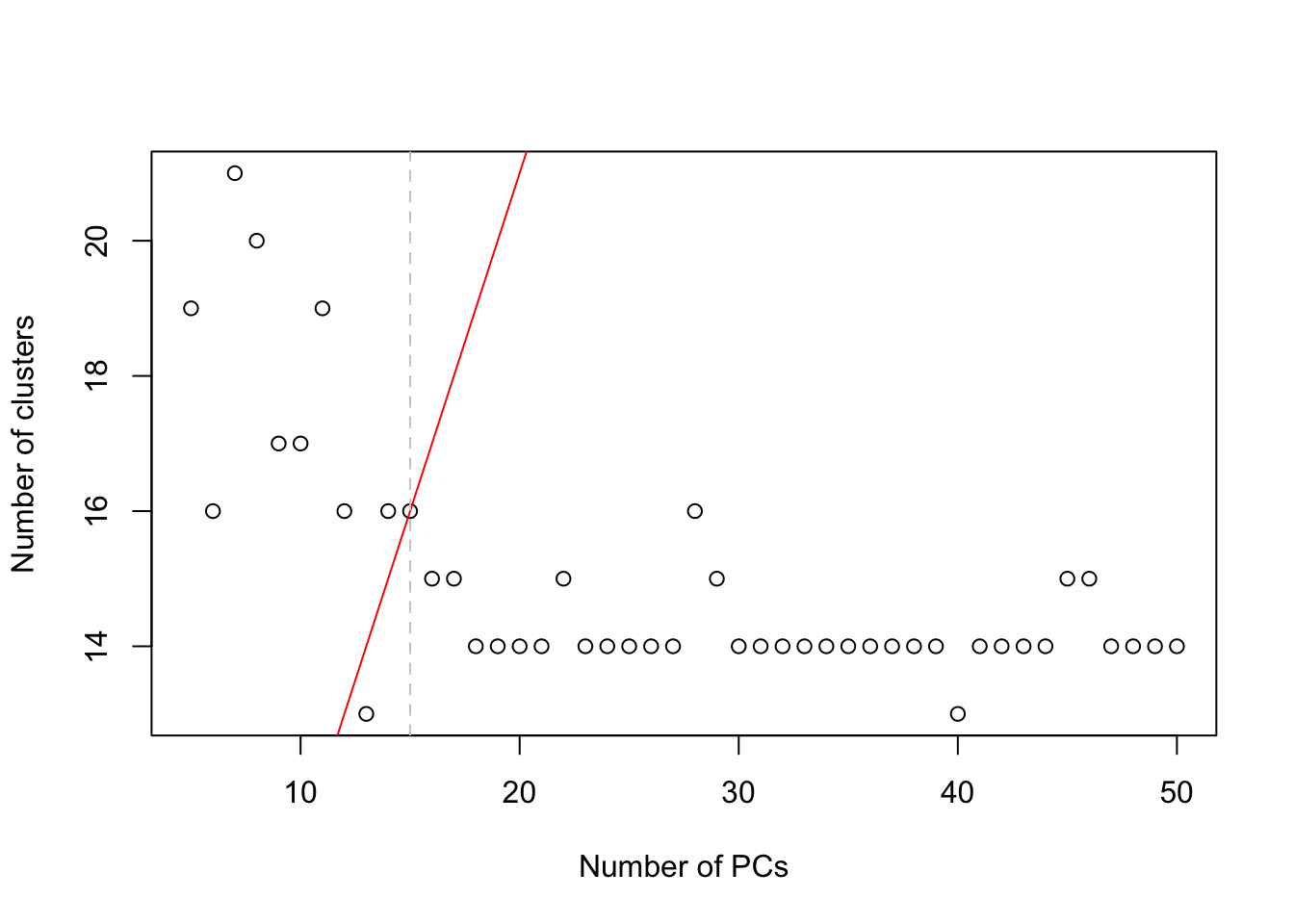
Supongamos que esperamos d subpoblaciones de células, en ese caso, necesitamos d-1 dimensiones para garantizar la separación de todas las subpoblaciones.
Pero… en un escenario real realmente no sabes cuántas poblaciones hay…
- Intenta con un rango para d y únicamente considera valores que produzcan a lo más d+1 clusters
- Cuando se seleccionan más clusters con menos dimensiones se produce ‘overclustering’.
- Elige una d que maximice el número de clusters sin caer en ‘overclustering’.
Ventaja: Es una solución pragmática que soluciona el equilibrio sesgo-varianza en los análisis posteriores (especialmente en el análisis de clustering).
Desventaja: Hace suposiciones fuertes sobre la naturaleza de las diferencias biológicas entre los clusters, y de hecho supone la existencia de clusters, los cuales podrían no existir en algunos procesos biológicos como la diferenciación.
5.6.4 Juntando todo
set.seed(100)
# Calcula y almacena todos los posibles PCs
sce.zeisel <- runPCA(sce.zeisel, subset_row = top.zeisel)
# Selecciona y almacena el conjunto reducido de PCs:
# ... por el método del codo
reducedDim(sce.zeisel, "PCA_elbow") <- reducedDim(
sce.zeisel, "PCA"
)[, 1:chosen.elbow]
# ... basado en la estructura poblacional
reducedDim(sce.zeisel, "PCA_clusters") <- reducedDim(
sce.zeisel, "PCA"
)[, 1:chosen.clusters]5.6.5 EJERCICIO
- Realiza un PCA para los datos sce.pbmc.
- Elige el número de PCs a conservar utilizando el método del codo.
- Elige el número de PCs a conservar utilizando la estructura de la población.
- Agrega esta información al objeto sce.pbmc.
5.6.6 Usando el ruido técnico
Otra técnica de reducción de dimensiones consiste en conservar todos los PCs hasta que el % de variación explicado alcance algun límite (por ejemplo, basado en la estimación de la variación técnica).
denoisePCA() automáticamente selecciona el número de PCs.
Por default, denoisePCA() realiza algunas simulaciones, por lo tanto necesitamos ‘configurar la semilla’ para obtener resultados reproducibles.
library(scran)
set.seed(111001001)
denoised.pbmc <- denoisePCA(sce.pbmc,
technical = dec.pbmc, subset.row = top.pbmc
)## [1] 3968 8La dimensionalidad del output es el límite inferior para el número de PCs requeridos para explicar toda la variación biológica. Lo que significa que cualquier número menor de PCs definitivamente descartará algún aspecto de la señal biológica.
Esto no grantiza que los PCs retenidos capturen toda la señal biológica
Esta técnica usualmente retiene más PCs que el método del punto del codo
scran::denoisePCA() internamente limita el numero de PCs, por default 5-50, para evitar la selección de excesivamente pocos PCs (cuando el ruido técnico es alto relativo al ruido biológico) o excesivamente muchos PCs (cuando el ruido técnico es demasiado bajo).
5.6.6.1 ¿Qué pasa si calculamos la relación media-varianza con la función modelGeneVar para el dataset sce.pbmc (anteriormente usamos la función modelGeneVarByPoisson para este propósito)?
dec.pbmc2 <- modelGeneVar(sce.pbmc)
denoised.pbmc2 <- denoisePCA(sce.pbmc,
technical = dec.pbmc2, subset.row = top.pbmc
)
dim(reducedDim(denoised.pbmc2))## [1] 3968 5scran::denoisePCA() tiende a funcionar mejor cuando la relación media-varianza refleja el ruido técnico verdadero, i.e estimado por scran::modelGeneVarByPoisson() o scran::modelGeneVarWithSpikes() en vez de scran::modelGeneVar().
El dataset PBMC está cerca de este límite inferior: el ruido técnico es alto relativo al ruido biológico.
5.6.6.2 ¿Qué pasa si calculamos el número de PCs usando el ruido técnico para el dataset sce.zeisel?
set.seed(001001001)
denoised.zeisel <- denoisePCA(sce.zeisel,
technical = dec.zeisel, subset.row = top.zeisel
)
dim(reducedDim(denoised.zeisel))## [1] 2815 50Los datos de cerebro de Zeisel están cerca de este límite superior: el ruido técnico es demasiado bajo
## [1] 2815 50## [1] 2815 7## [1] 2815 155.7 Reducción de dimensionalidad para visualización
5.7.1 Motivación
Los algoritmos de clustering, así como la mayoría de los algoritmos, operan fácilmente sobre 10-50 (a lo más) PCs, pero ese número es aún demasiado para la visualización.
Por lo tanto, necesitamos estrategias adicionales para la reducción de dimensionalidad si queremos visualizar los datos.
5.7.2 Visualizando con PCA
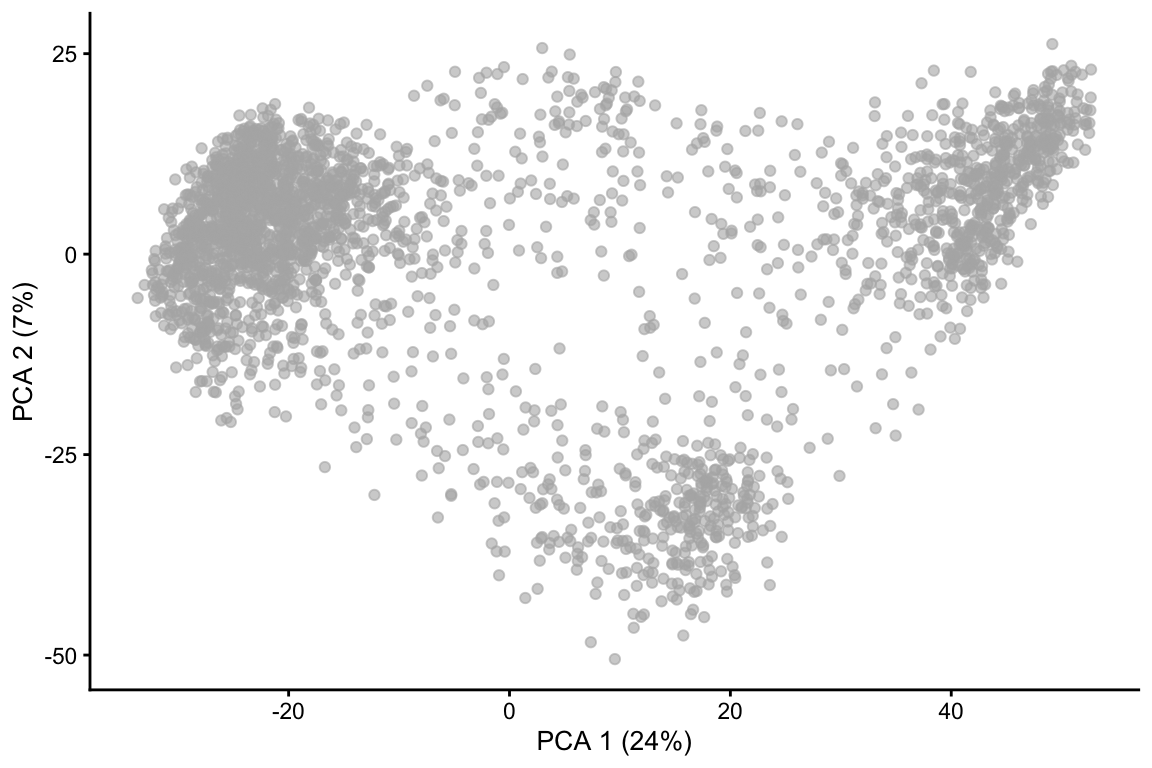
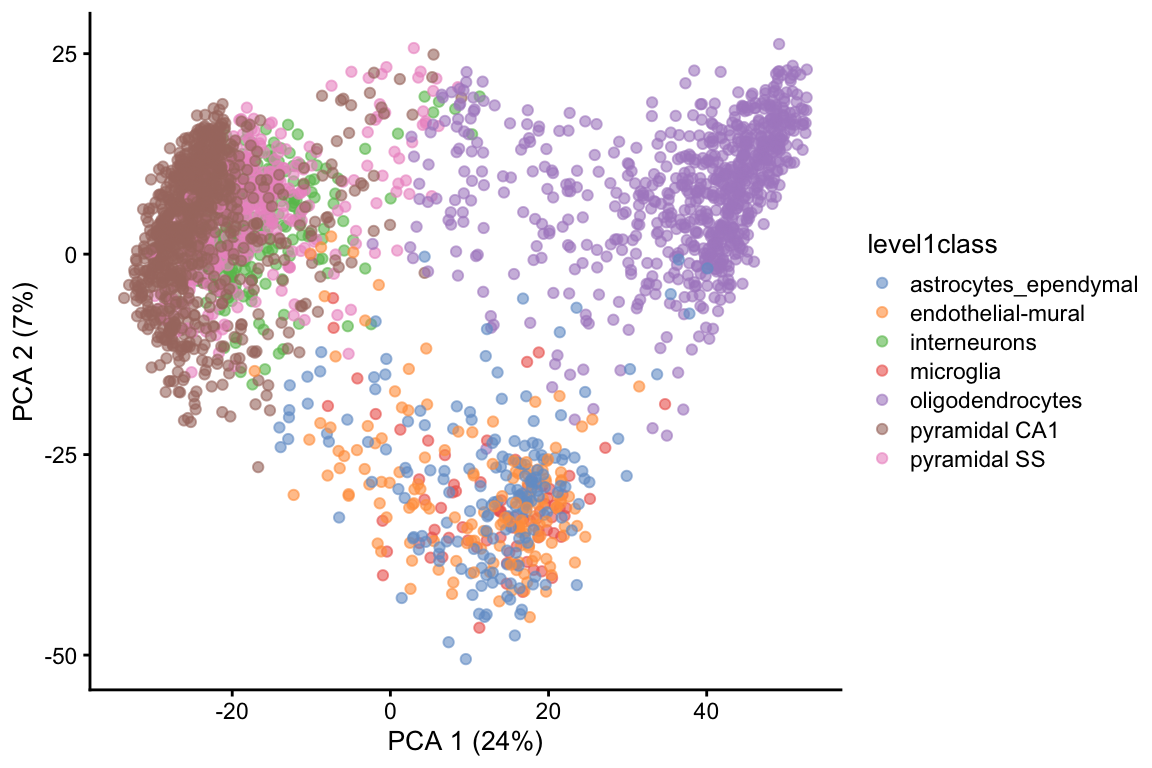
PCA es una técnica lineal, por lo tanto, no es eficiente para comprimir diferencias en más de 2 dimensiones en los primeros 2 PCs.
5.7.3 Retos y resumen de la visualización con PCA
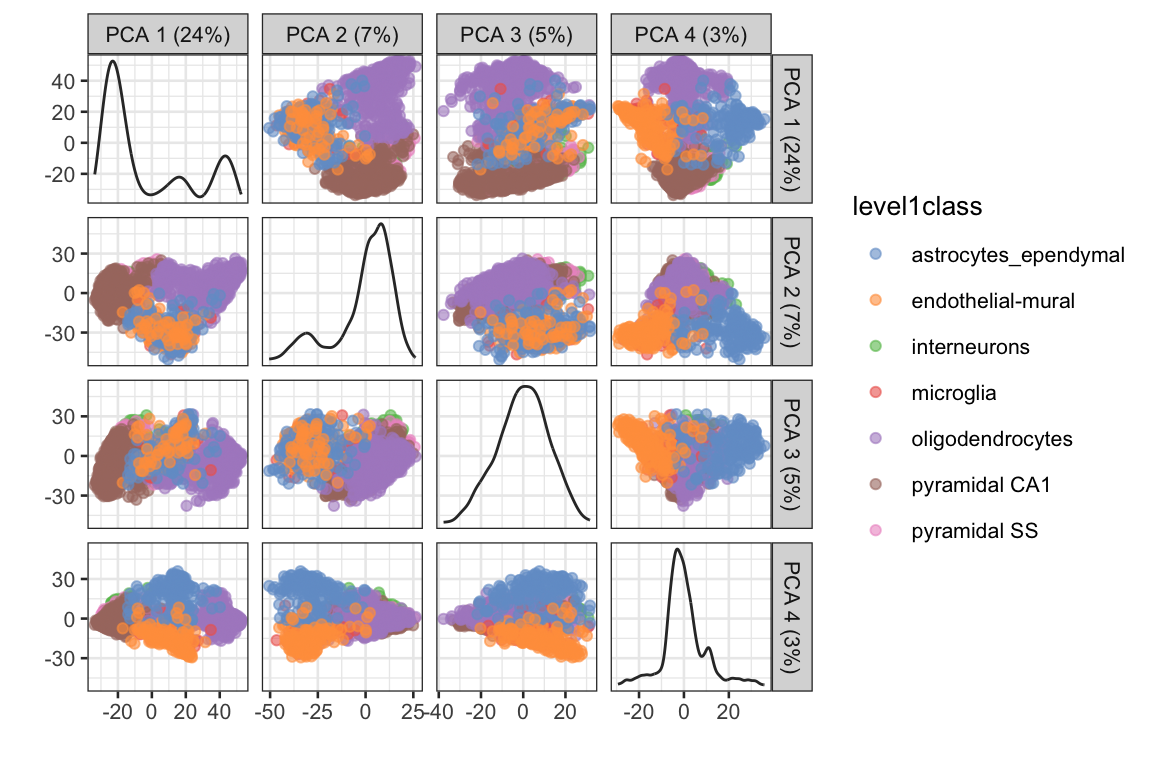
Ventajas:
- PCA es predecible y no introducirá estructura artificial en los datos.
- Es determínistico y robusto a cambios pequeños en los valores de entrada.
Desventajas:
- Usualmente la visualización no es suficiente para visualizar la naturaleza compleja de los datos de scRNA-seq.
5.7.4 Visualización con t-SNE
set.seed(00101001101)
sce.zeisel <- runTSNE(sce.zeisel, dimred = "PCA")
plotReducedDim(sce.zeisel, dimred = "TSNE", colour_by = "level1class")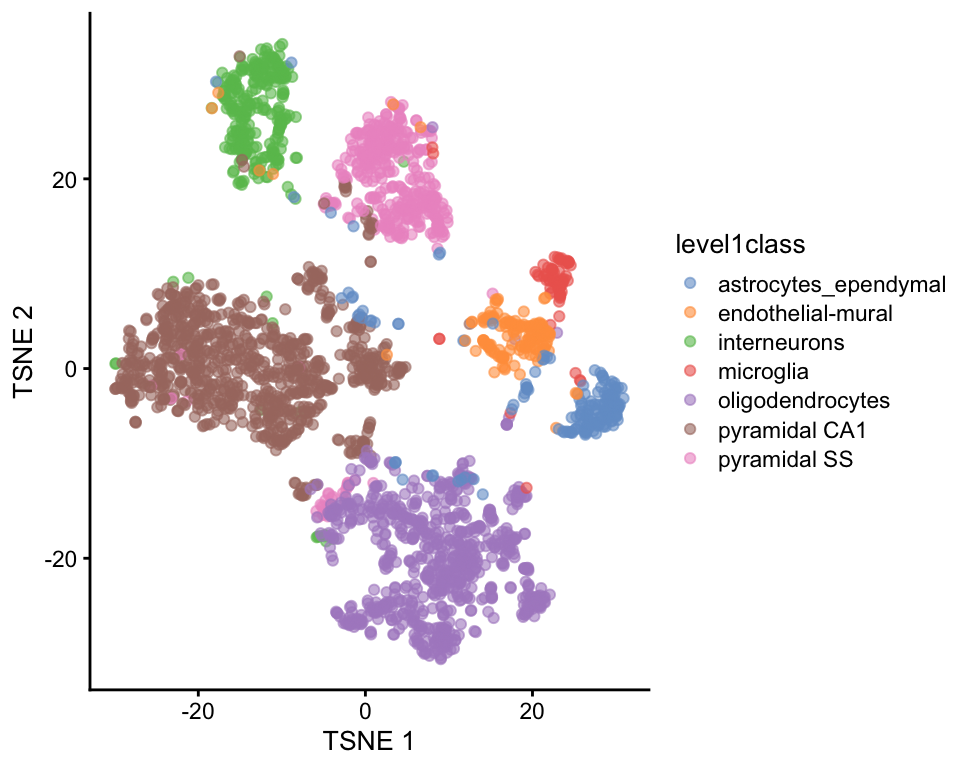
t-stochastic neighbour embedding (t-SNE) es la visualización por excelencia de datos de scRNA-seq. Intenta encontrar una representación (no-lineal) de los datos usando pocas dimensiones que preserve las distancias entre cada punto y sus vecinos en el espacio multi-dimensional
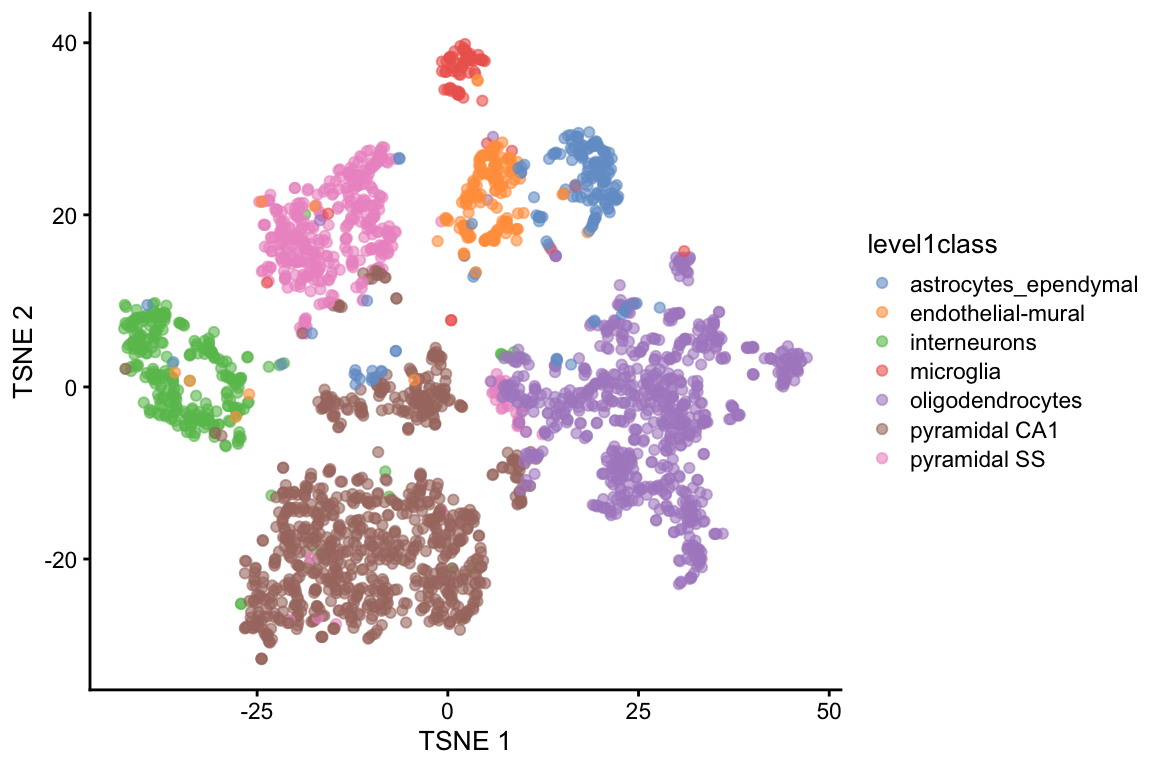
5.7.4.1 Retos de la visualización con t-SNE
set.seed(100)
sce.zeisel <- runTSNE(sce.zeisel,
dimred = "PCA",
perplexity = 30
)
plotReducedDim(sce.zeisel,
dimred = "TSNE",
colour_by = "level1class"
)
5.7.4.2 Preguntas
¿Qué pasa si vuelves a correr runTSNE() sin especificar la semilla?
¿Qué pasa si especificas la semilla pero cambias el valor del parámetro perplexity?
5.7.4.3 Continuando
- Baja perplejidad favorece la resolución de la estructura fina, posiblemente al grado de que la visualización parece ruido random.
set.seed(100)
sce.zeisel <- runTSNE(sce.zeisel, dimred = "PCA", perplexity = 5)
p1 <- plotReducedDim(sce.zeisel, dimred = "TSNE", colour_by = "level1class")
sce.zeisel <- runTSNE(sce.zeisel, dimred = "PCA", perplexity = 20)
p2 <- plotReducedDim(sce.zeisel, dimred = "TSNE", colour_by = "level1class")
sce.zeisel <- runTSNE(sce.zeisel, dimred = "PCA", perplexity = 80)
p3 <- plotReducedDim(sce.zeisel, dimred = "TSNE", colour_by = "level1class")
library("patchwork")
p1 + p2 + p3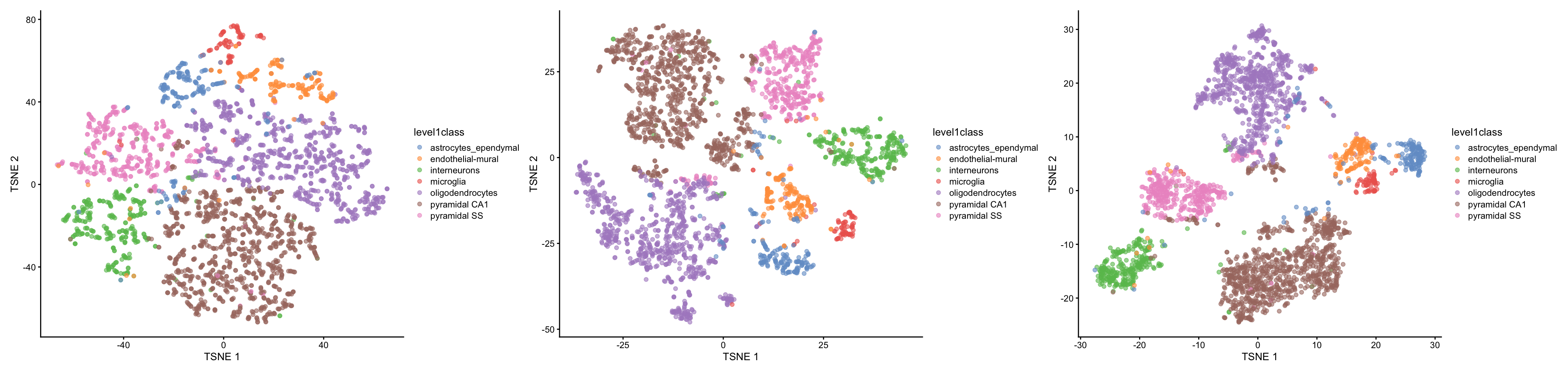
El siguiente foro discute la selección de parámetros para t-SNE con cierta profundidad.
- No sobreinterpretes los resultados de t-SNE como un ‘mapa’ de las identidades de las células individuales.
- Algunos componentes aleatorios y la selección de parámetros cambiarán la visualización.
- La interpretación puede ser engañada por el tamaño y posición de los clusters.
- t-SNE infla clusters densos y comprime clusters escasos.
- t-SNE no está obligado a preservar las localizaciones relativas de clusters no-vecinos (no puedes interpretar distancias no locales).
Aún así: t-SNE es una herramienta probada para visualización general de datos de scRNA-seq y sigue siendo muy popular
5.7.5 Visualización con UMAP
Uniform manifold approximation and project (UMAP) es una alternativa a t-SNE.
Así como t-SNE, UMAP intenta encontrar una representación (no lineal) de pocas dimensiones de los datos que preserve las distancias entre cada punto y sus vecinos en el espacio multi-dimensional.
t-SNE y UMAP están basados en diferentes teorías matemáticas.
set.seed(1100101001)
sce.zeisel <- runUMAP(sce.zeisel, dimred = "PCA")
plotReducedDim(sce.zeisel,
dimred = "UMAP",
colour_by = "level1class"
)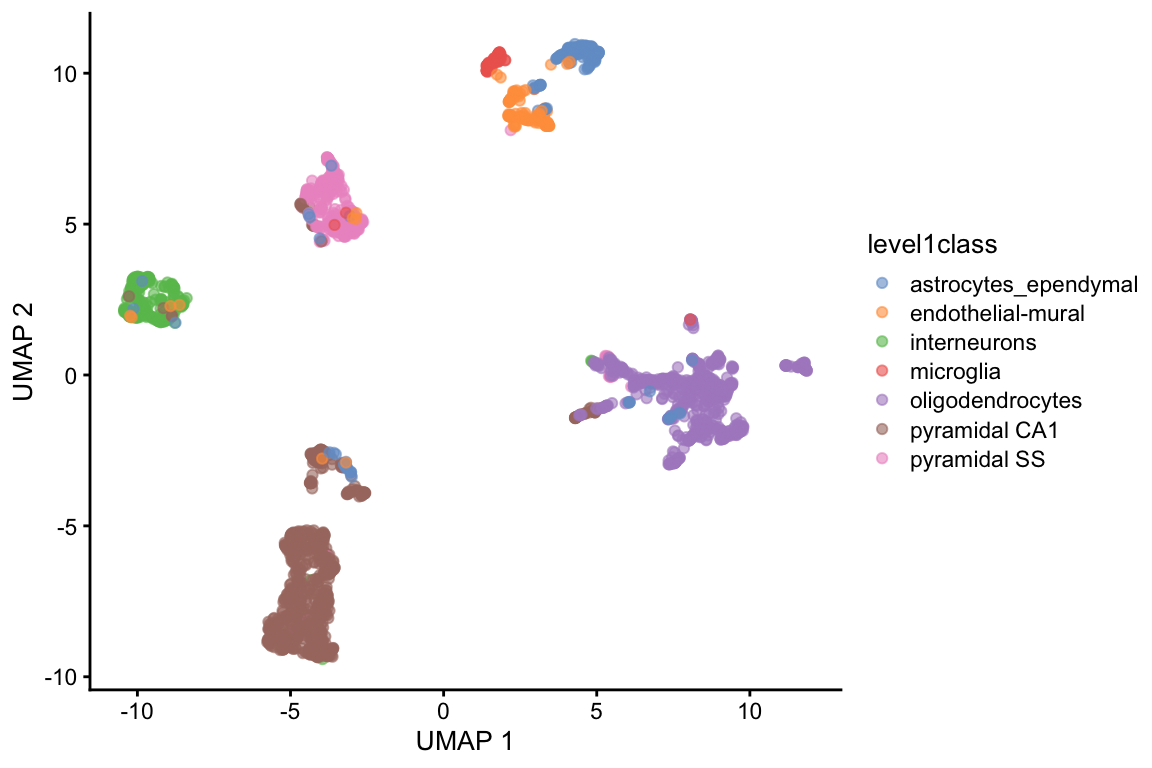
Comparado con t-SNE:
- UMAP tiende a encontrar clusters visualmente más compactos.
- Intenta preservar más de la estructura global que t-SNE.
- Tiende a ser más rápido que t-SNE, lo cual puede ser importante para datasets grandes. La diferencia desaparece cuando se aplican sobre los primeros PCs.
5.7.5.1 Preguntas
¿Qué pasa si vuelves a correr runUMAP() sin especificar la semilla?
¿Qué pasa si especificas la semilla pero cambias el valor del parámetro n_neighbors?
5.7.5.2 Continuando
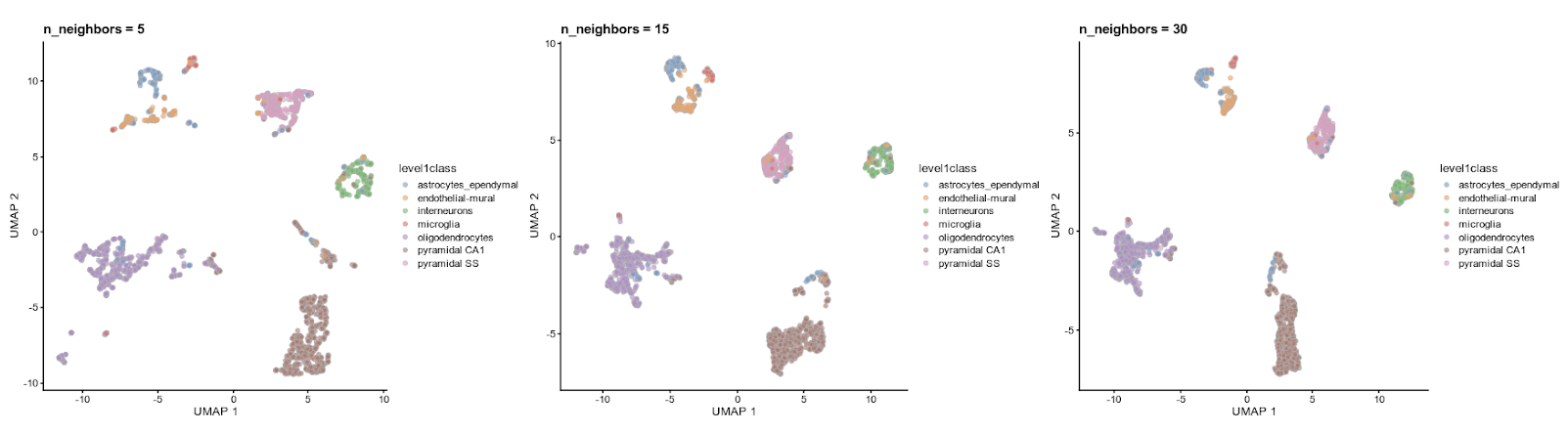
Igual que para t-SNE, es necesario configurar una semilla y diferentes valores para los parámetros cambiaron la visualización.
Si el valor para los parámetros n_neighbors o min_dist es demasiado bajo entonces el ruido aleatorio se interpretará como estructura de alta-resolución, si son demasiado altos entonces se perderá la estructura fina.
TIP: Trata un rango de valores para cada parámetro para asegurarte de que no comprometen ninguna de las conclusiones derivadas de la gráfica UMAP o t-SNE
5.7.6 Interpretando las gráficas
Recuerda:
Reducción de dimensionalidad para la visualización de los datos necesariamente involucra descartar información y distorsionar las distancias entre las células.
No sobre interpretes las gráficas bonitas.
5.7.7 Resumen y recomendaciones
Las gráficas de t-SNE y UMAP son herramientas diagnóstico importantes, por ejemplo: para checar si dos clusters son realmente subclusters vecinos o si un cluster puede ser dividido en más de un cluster.
Es debatible cuál visualización, t-SNE o UMAP, es más útil o estéticamente agradable.
Está bien elegir aquella que funcione mejor para tu análisis (tomando en cuenta que tratarás la gráfica únicamente como una herramienta de visualización/diagnóstico y que no llegarás a ninguna conclusión fuerte basado únicamente en la gráfica).
5.9 Detalles de la sesión de R
## [1] "2023-08-10 10:12:22 EDT"## user system elapsed
## 1940.654 132.562 136575.205## ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
## setting value
## version R version 4.3.1 (2023-06-16)
## os macOS Ventura 13.4.1
## system aarch64, darwin20
## ui RStudio
## language (EN)
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz America/New_York
## date 2023-08-10
## rstudio 2023.06.0+421 Mountain Hydrangea (desktop)
## pandoc 3.1.1 @ /Applications/RStudio.app/Contents/Resources/app/quarto/bin/tools/ (via rmarkdown)
##
## ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
## package * version date (UTC) lib source
## abind 1.4-5 2016-07-21 [1] CRAN (R 4.3.0)
## AnnotationDbi * 1.62.2 2023-07-02 [1] Bioconductor
## AnnotationFilter * 1.24.0 2023-05-08 [1] Bioconductor
## AnnotationHub * 3.8.0 2023-05-08 [1] Bioconductor
## beachmat 2.16.0 2023-05-08 [1] Bioconductor
## beeswarm 0.4.0 2021-06-01 [1] CRAN (R 4.3.0)
## Biobase * 2.60.0 2023-05-08 [1] Bioconductor
## BiocFileCache * 2.8.0 2023-05-08 [1] Bioconductor
## BiocGenerics * 0.46.0 2023-06-04 [1] Bioconductor
## BiocIO 1.10.0 2023-05-08 [1] Bioconductor
## BiocManager 1.30.21.1 2023-07-18 [1] CRAN (R 4.3.0)
## BiocNeighbors 1.18.0 2023-05-08 [1] Bioconductor
## BiocParallel 1.34.2 2023-05-28 [1] Bioconductor
## BiocSingular 1.16.0 2023-05-08 [1] Bioconductor
## BiocVersion 3.17.1 2022-12-20 [1] Bioconductor
## biomaRt 2.56.1 2023-06-11 [1] Bioconductor
## Biostrings 2.68.1 2023-05-21 [1] Bioconductor
## bit 4.0.5 2022-11-15 [1] CRAN (R 4.3.0)
## bit64 4.0.5 2020-08-30 [1] CRAN (R 4.3.0)
## bitops 1.0-7 2021-04-24 [1] CRAN (R 4.3.0)
## blob 1.2.4 2023-03-17 [1] CRAN (R 4.3.0)
## bluster * 1.10.0 2023-05-08 [1] Bioconductor
## bookdown 0.34 2023-05-09 [1] CRAN (R 4.3.0)
## bslib 0.5.0 2023-06-09 [1] CRAN (R 4.3.0)
## cachem 1.0.8 2023-05-01 [1] CRAN (R 4.3.0)
## cli 3.6.1 2023-03-23 [1] CRAN (R 4.3.0)
## cluster 2.1.4 2022-08-22 [1] CRAN (R 4.3.1)
## codetools 0.2-19 2023-02-01 [1] CRAN (R 4.3.1)
## colorspace 2.1-0 2023-01-23 [1] CRAN (R 4.3.0)
## cowplot 1.1.1 2020-12-30 [1] CRAN (R 4.3.0)
## crayon 1.5.2 2022-09-29 [1] CRAN (R 4.3.0)
## curl 5.0.1 2023-06-07 [1] CRAN (R 4.3.0)
## DBI 1.1.3 2022-06-18 [1] CRAN (R 4.3.0)
## dbplyr * 2.3.3 2023-07-07 [1] CRAN (R 4.3.0)
## DelayedArray 0.26.7 2023-07-28 [1] Bioconductor
## DelayedMatrixStats 1.22.1 2023-06-09 [1] Bioconductor
## digest 0.6.33 2023-07-07 [1] CRAN (R 4.3.0)
## dplyr * 1.1.2 2023-04-20 [1] CRAN (R 4.3.0)
## dqrng 0.3.0 2021-05-01 [1] CRAN (R 4.3.0)
## DropletUtils * 1.20.0 2023-05-08 [1] Bioconductor
## edgeR 3.42.4 2023-06-04 [1] Bioconductor
## ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.3.0)
## EnsDb.Hsapiens.v86 * 2.99.0 2023-07-29 [1] Bioconductor
## ensembldb * 2.24.0 2023-05-08 [1] Bioconductor
## evaluate 0.21 2023-05-05 [1] CRAN (R 4.3.0)
## ExperimentHub 2.8.1 2023-07-16 [1] Bioconductor
## fansi 1.0.4 2023-01-22 [1] CRAN (R 4.3.0)
## farver 2.1.1 2022-07-06 [1] CRAN (R 4.3.0)
## fastmap 1.1.1 2023-02-24 [1] CRAN (R 4.3.0)
## filelock 1.0.2 2018-10-05 [1] CRAN (R 4.3.0)
## FNN 1.1.3.2 2023-03-20 [1] CRAN (R 4.3.0)
## generics 0.1.3 2022-07-05 [1] CRAN (R 4.3.0)
## GenomeInfoDb * 1.36.1 2023-07-02 [1] Bioconductor
## GenomeInfoDbData 1.2.10 2023-06-08 [1] Bioconductor
## GenomicAlignments 1.36.0 2023-05-08 [1] Bioconductor
## GenomicFeatures * 1.52.1 2023-07-02 [1] Bioconductor
## GenomicRanges * 1.52.0 2023-05-08 [1] Bioconductor
## ggbeeswarm 0.7.2 2023-04-29 [1] CRAN (R 4.3.0)
## ggplot2 * 3.4.2 2023-04-03 [1] CRAN (R 4.3.0)
## ggrepel * 0.9.3 2023-02-03 [1] CRAN (R 4.3.0)
## glue 1.6.2 2022-02-24 [1] CRAN (R 4.3.0)
## gridExtra 2.3 2017-09-09 [1] CRAN (R 4.3.0)
## gtable 0.3.3 2023-03-21 [1] CRAN (R 4.3.0)
## HDF5Array 1.28.1 2023-05-08 [1] Bioconductor
## here 1.0.1 2020-12-13 [1] CRAN (R 4.3.0)
## highr 0.10 2022-12-22 [1] CRAN (R 4.3.0)
## hms 1.1.3 2023-03-21 [1] CRAN (R 4.3.0)
## htmltools 0.5.5 2023-03-23 [1] CRAN (R 4.3.0)
## httpuv 1.6.11 2023-05-11 [1] CRAN (R 4.3.0)
## httr 1.4.6 2023-05-08 [1] CRAN (R 4.3.0)
## igraph 1.5.0.1 2023-07-23 [1] CRAN (R 4.3.0)
## interactiveDisplayBase 1.38.0 2023-05-08 [1] Bioconductor
## IRanges * 2.34.1 2023-07-02 [1] Bioconductor
## irlba 2.3.5.1 2022-10-03 [1] CRAN (R 4.3.0)
## jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.3.0)
## jsonlite 1.8.7 2023-06-29 [1] CRAN (R 4.3.0)
## kableExtra * 1.3.4 2021-02-20 [1] CRAN (R 4.3.0)
## KEGGREST 1.40.0 2023-05-08 [1] Bioconductor
## knitr 1.43 2023-05-25 [1] CRAN (R 4.3.0)
## labeling 0.4.2 2020-10-20 [1] CRAN (R 4.3.0)
## later 1.3.1 2023-05-02 [1] CRAN (R 4.3.0)
## lattice 0.21-8 2023-04-05 [1] CRAN (R 4.3.1)
## lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.3.0)
## lifecycle 1.0.3 2022-10-07 [1] CRAN (R 4.3.0)
## limma 3.56.2 2023-06-04 [1] Bioconductor
## locfit 1.5-9.8 2023-06-11 [1] CRAN (R 4.3.0)
## magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.3.0)
## Matrix * 1.6-0 2023-07-08 [1] CRAN (R 4.3.0)
## MatrixGenerics * 1.12.3 2023-07-30 [1] Bioconductor
## matrixStats * 1.0.0 2023-06-02 [1] CRAN (R 4.3.0)
## memoise 2.0.1 2021-11-26 [1] CRAN (R 4.3.0)
## metapod 1.8.0 2023-04-25 [1] Bioconductor
## mime 0.12 2021-09-28 [1] CRAN (R 4.3.0)
## munsell 0.5.0 2018-06-12 [1] CRAN (R 4.3.0)
## patchwork * 1.1.2 2022-08-19 [1] CRAN (R 4.3.0)
## PCAtools * 2.12.0 2023-05-08 [1] Bioconductor
## pheatmap * 1.0.12 2019-01-04 [1] CRAN (R 4.3.0)
## pillar 1.9.0 2023-03-22 [1] CRAN (R 4.3.0)
## pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.3.0)
## plyr 1.8.8 2022-11-11 [1] CRAN (R 4.3.0)
## png 0.1-8 2022-11-29 [1] CRAN (R 4.3.0)
## prettyunits 1.1.1 2020-01-24 [1] CRAN (R 4.3.0)
## progress 1.2.2 2019-05-16 [1] CRAN (R 4.3.0)
## promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.3.0)
## ProtGenerics 1.32.0 2023-05-08 [1] Bioconductor
## purrr 1.0.1 2023-01-10 [1] CRAN (R 4.3.0)
## R.methodsS3 1.8.2 2022-06-13 [1] CRAN (R 4.3.0)
## R.oo 1.25.0 2022-06-12 [1] CRAN (R 4.3.0)
## R.utils 2.12.2 2022-11-11 [1] CRAN (R 4.3.0)
## R6 2.5.1 2021-08-19 [1] CRAN (R 4.3.0)
## rappdirs 0.3.3 2021-01-31 [1] CRAN (R 4.3.0)
## RColorBrewer 1.1-3 2022-04-03 [1] CRAN (R 4.3.0)
## Rcpp 1.0.11 2023-07-06 [1] CRAN (R 4.3.0)
## RCurl 1.98-1.12 2023-03-27 [1] CRAN (R 4.3.0)
## reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.3.0)
## restfulr 0.0.15 2022-06-16 [1] CRAN (R 4.3.0)
## rhdf5 2.44.0 2023-05-08 [1] Bioconductor
## rhdf5filters 1.12.1 2023-05-08 [1] Bioconductor
## Rhdf5lib 1.22.0 2023-05-08 [1] Bioconductor
## rjson 0.2.21 2022-01-09 [1] CRAN (R 4.3.0)
## rlang 1.1.1 2023-04-28 [1] CRAN (R 4.3.0)
## rmarkdown 2.23 2023-07-01 [1] CRAN (R 4.3.0)
## rprojroot 2.0.3 2022-04-02 [1] CRAN (R 4.3.0)
## Rsamtools 2.16.0 2023-06-04 [1] Bioconductor
## RSQLite 2.3.1 2023-04-03 [1] CRAN (R 4.3.0)
## rstudioapi 0.15.0 2023-07-07 [1] CRAN (R 4.3.0)
## rsvd 1.0.5 2021-04-16 [1] CRAN (R 4.3.0)
## rtracklayer 1.60.0 2023-05-08 [1] Bioconductor
## Rtsne 0.16 2022-04-17 [1] CRAN (R 4.3.0)
## rvest 1.0.3 2022-08-19 [1] CRAN (R 4.3.0)
## S4Arrays 1.0.5 2023-07-24 [1] Bioconductor
## S4Vectors * 0.38.1 2023-05-08 [1] Bioconductor
## sass 0.4.7 2023-07-15 [1] CRAN (R 4.3.0)
## ScaledMatrix 1.8.1 2023-05-08 [1] Bioconductor
## scales 1.2.1 2022-08-20 [1] CRAN (R 4.3.0)
## scater * 1.28.0 2023-04-25 [1] Bioconductor
## scran * 1.28.2 2023-07-23 [1] Bioconductor
## scRNAseq * 2.14.0 2023-04-27 [1] Bioconductor
## scuttle * 1.9.4 2023-01-23 [1] Bioconductor
## sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.3.0)
## shiny 1.7.4.1 2023-07-06 [1] CRAN (R 4.3.0)
## SingleCellExperiment * 1.22.0 2023-05-08 [1] Bioconductor
## sparseMatrixStats 1.12.2 2023-07-02 [1] Bioconductor
## statmod 1.5.0 2023-01-06 [1] CRAN (R 4.3.0)
## stringi 1.7.12 2023-01-11 [1] CRAN (R 4.3.0)
## stringr 1.5.0 2022-12-02 [1] CRAN (R 4.3.0)
## SummarizedExperiment * 1.30.2 2023-06-06 [1] Bioconductor
## svglite 2.1.1 2023-01-10 [1] CRAN (R 4.3.0)
## systemfonts 1.0.4 2022-02-11 [1] CRAN (R 4.3.0)
## tibble 3.2.1 2023-03-20 [1] CRAN (R 4.3.0)
## tidyselect 1.2.0 2022-10-10 [1] CRAN (R 4.3.0)
## utf8 1.2.3 2023-01-31 [1] CRAN (R 4.3.0)
## uwot 0.1.16 2023-06-29 [1] CRAN (R 4.3.0)
## vctrs 0.6.3 2023-06-14 [1] CRAN (R 4.3.0)
## vipor 0.4.5 2017-03-22 [1] CRAN (R 4.3.0)
## viridis 0.6.4 2023-07-22 [1] CRAN (R 4.3.0)
## viridisLite 0.4.2 2023-05-02 [1] CRAN (R 4.3.0)
## webshot 0.5.5 2023-06-26 [1] CRAN (R 4.3.0)
## withr 2.5.0 2022-03-03 [1] CRAN (R 4.3.0)
## xfun 0.39 2023-04-20 [1] CRAN (R 4.3.0)
## XML 3.99-0.14 2023-03-19 [1] CRAN (R 4.3.0)
## xml2 1.3.5 2023-07-06 [1] CRAN (R 4.3.0)
## xtable 1.8-4 2019-04-21 [1] CRAN (R 4.3.0)
## XVector 0.40.0 2023-05-08 [1] Bioconductor
## yaml 2.3.7 2023-01-23 [1] CRAN (R 4.3.0)
## zlibbioc 1.46.0 2023-05-08 [1] Bioconductor
##
## [1] /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/library
##
## ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────