3 Normalización de datos
Instructora: Dra Laura Lucila Gómez Romero
3.2 Motivación
Al igual que con otras tecnologías, single-cell RNA-seq (scRNA-seq) puede presentar diferencias sistemáticas en la cobertura de las librerías de scRAN-seq.
Típicamente se debe a diferencias técnicas en procesos como la captura de cDNA, la eficiencia de la amplificación por PCR.
La normalización tiene como objetivo remover estas diferencias sistemáticas para que no interfieran cuando comparamos los perfiles de expresión entre células.
Al normalizar los datos, las diferencias observadas entre poblaciones celulares o condiciones son debido a la biología y no por factores técnicos.
La normalización es diferente a la corrección por lote . El proceso de normalización se lleva a cabo sin importar la estructura de los lotes y sólo considera sesgos técnicos, los cuales tienden a afectar los genes en una manera medianamente similar, por ejemplo: la eficiencia de la amplificación por PCR debido al contenido de GC o la longitud del gene.
- ¿Qué es correción por lote (batch effect correction)? Da un ejemplo.
“Large single-cell RNA sequencing (scRNA-seq) projects usually need to generate data across multiple batches due to logistical constraints. However, the processing of different batches is often subject to uncontrollable differences, e.g., changes in operator, differences in reagent quality. This results in systematic differences in the observed expression in cells from different batches, which we refer to as “batch effects”. Batch effects are problematic as they can be major drivers of heterogeneity in the data, masking the relevant biological differences and complicating interpretation of the results” -OSCA
- ¿Cuáles son las diferencias entre correción por lote y normalización?
“Normalization occurs regardless of the batch structure and only considers technical biases, while batch correction - as the name suggests - only occurs across batches and must consider both technical biases and biological differences. Technical biases tend to affect genes in a similar manner, or at least in a manner related to their biophysical properties (e.g., length, GC content), while biological differences between batches can be highly unpredictable” -OSCA
3.3 Datos
Usaremos el dataset de Zeisel.
Tipos celulares en cerebro de ratón (oligodendrocitos, microglias, neuronas, etc.)
Procesado con STRT-seq (similar a CEL-seq), un sistema de microfluido.
3005 células y 18441 genes.
Contiene UMIs.
## class: SingleCellExperiment
## dim: 18441 3005
## metadata(0):
## assays(1): counts
## rownames(18441): ENSMUSG00000029669 ENSMUSG00000046982 ... ENSMUSG00000064337 ENSMUSG00000065947
## rowData names(2): featureType originalName
## colnames(3005): 1772071015_C02 1772071017_G12 ... 1772066098_A12 1772058148_F03
## colData names(10): tissue group # ... level1class level2class
## reducedDimNames(0):
## mainExpName: endogenous
## altExpNames(2): ERCC repeat3.3.1 Primero se debe hacer el control de calidad
Eliminando células de baja calidad:
Outliers bajos para log(cuenta total)
Outliers bajos para log(número de features detectados)
Outliers altos para el porcentaje de cuentas de conjuntos de genes especificados (mitocondriales, transcritos spike-in)
# Control de calidad
library("scater")
unfiltered <- sce.zeisel
is.mito <- which(rowData(sce.zeisel)$featureType == "mito")
stats <- perCellQCMetrics(sce.zeisel, subsets = list(Mt = is.mito))
qc <-
quickPerCellQC(stats,
percent_subsets = c("altexps_ERCC_percent", "subsets_Mt_percent")
)
colSums(as.data.frame(qc))## low_lib_size low_n_features high_altexps_ERCC_percent high_subsets_Mt_percent
## 0 3 66 128
## discard
## 1903.4 Normalización por escalamiento (scaling normalization)
La normalización por escalamiento es la estrategia más simple y usada.
Suposición: Cualquier sesgo específico en cada célula afecta a todos los genes de igual manera a través de escalar por el promedio esperado de cuentas para dicha célula.
Se realiza de la siguiente manera:
Estima un “size factor” para cada célula.
Este factor representa el sesgo relativo en esa célula.
Divide las cuentas de los genes de cada célula entre su correspondiente “size factor”.
\[ CuentasNormalizadas = Cuentas / Size factor\]
Los valores de expresión normalizados pueden ser usados por análisis posteriores como clustering o reducción de dimensiones.
3.4.1 Normalizacion por tamaño de biblioteca (Library Size normalization)
Tamaño de biblioteca (Library Size): La suma total de las cuentas a tráves de todos los genes en una célula. Asumimos que este valor escala con cualquier sesgo específico en cada célula.
\[Library Size_{cell} = \sum_{n=1}^{j} gene\] Donde \(j\) es el número total de genes y \(gene\) es el número de cuentas por gen en la célula \(cell\).
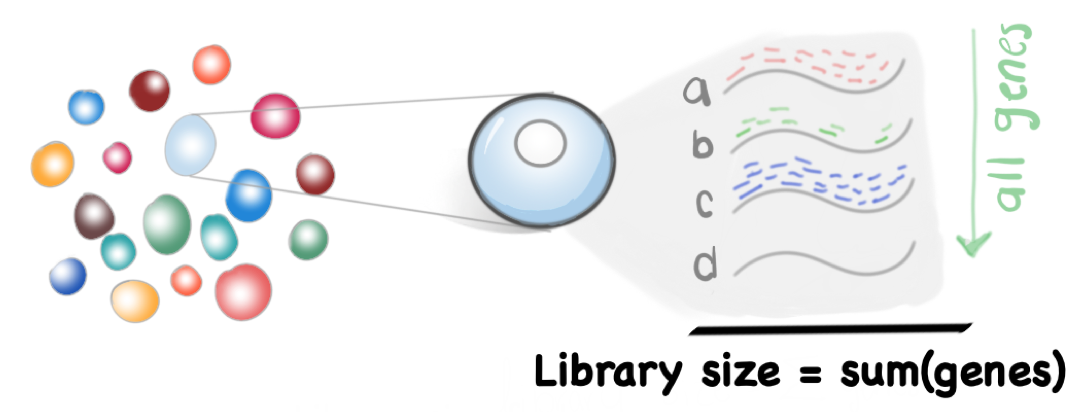
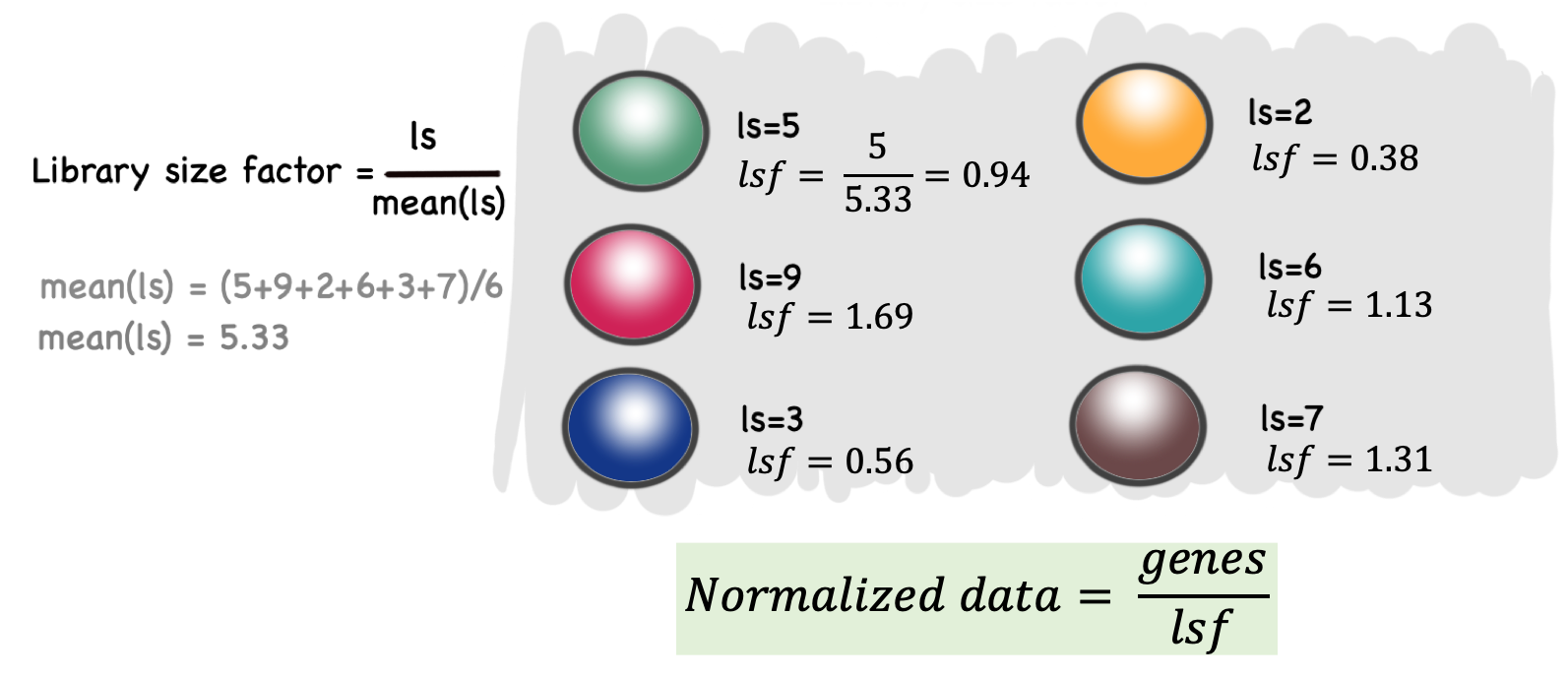
Para escalar los datos ocuparemos un factor de escalamiento llamado Library Size factor. Este valor se calcula a partir de escalar el tamaño de la biblioteca, tal que el promedio de los Library Size factor en todas las células sea igual a 1.
\[ mean(Library Size factor) = 1 \]
Lo que nos permite que los valores normalizados están en la misma escala y pueden ser útiles para la interpretación.
# Estimar factores de normalización
lib.sf.zeisel <- librarySizeFactors(sce.zeisel)
# Examina la distribución de los tamaños de librerías
# que acabamos de estimar
summary(lib.sf.zeisel)## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.1754 0.5682 0.8669 1.0000 1.2758 4.0651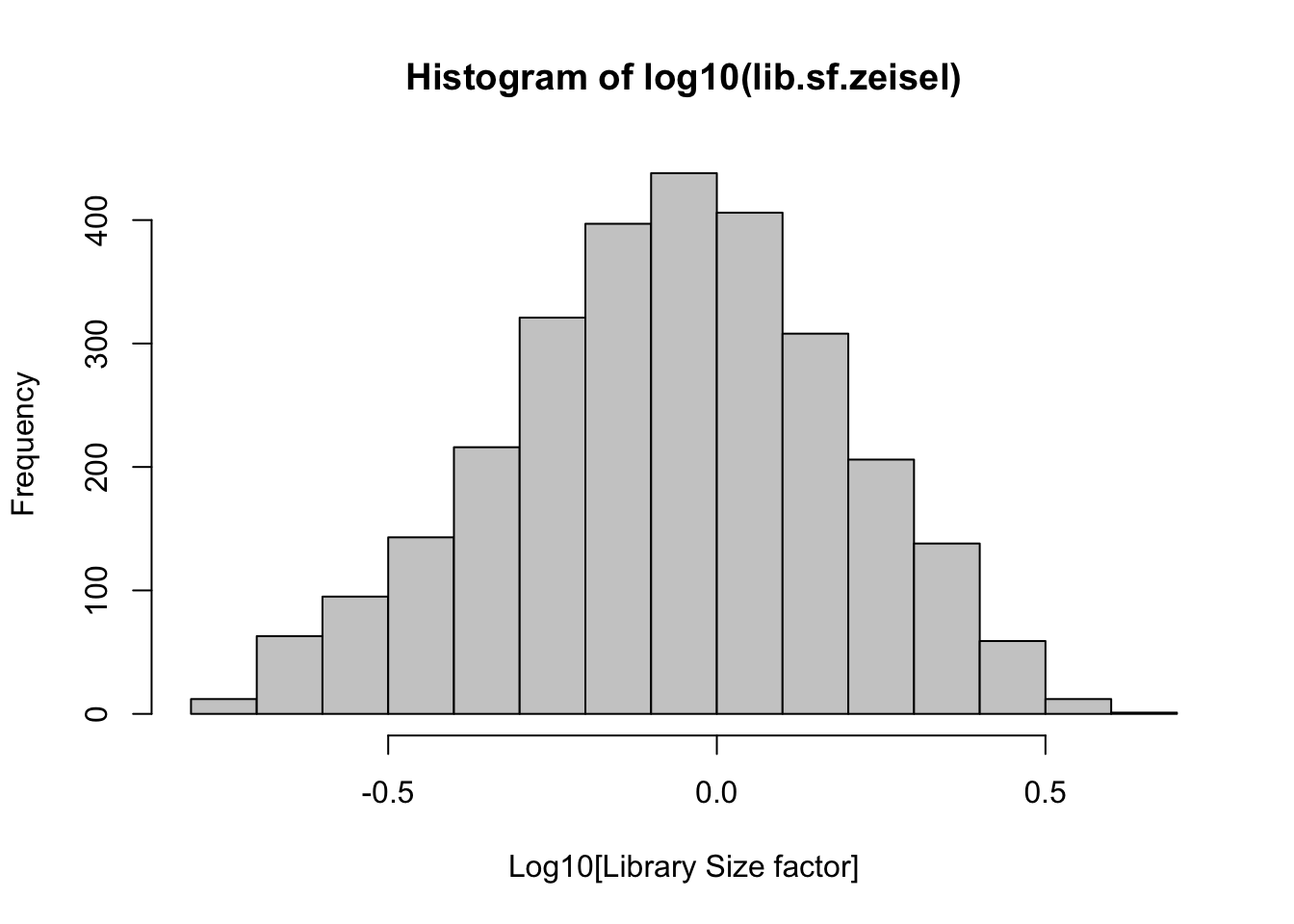
¿A qué crees que se deba la variación en los factores de escalamiento?
## Calculando el tamaño de las librerias
ls.zeisel <- colSums(counts(sce.zeisel))
plot(ls.zeisel, lib.sf.zeisel, log="xy",
xlab="Library size", ylab="Size factor")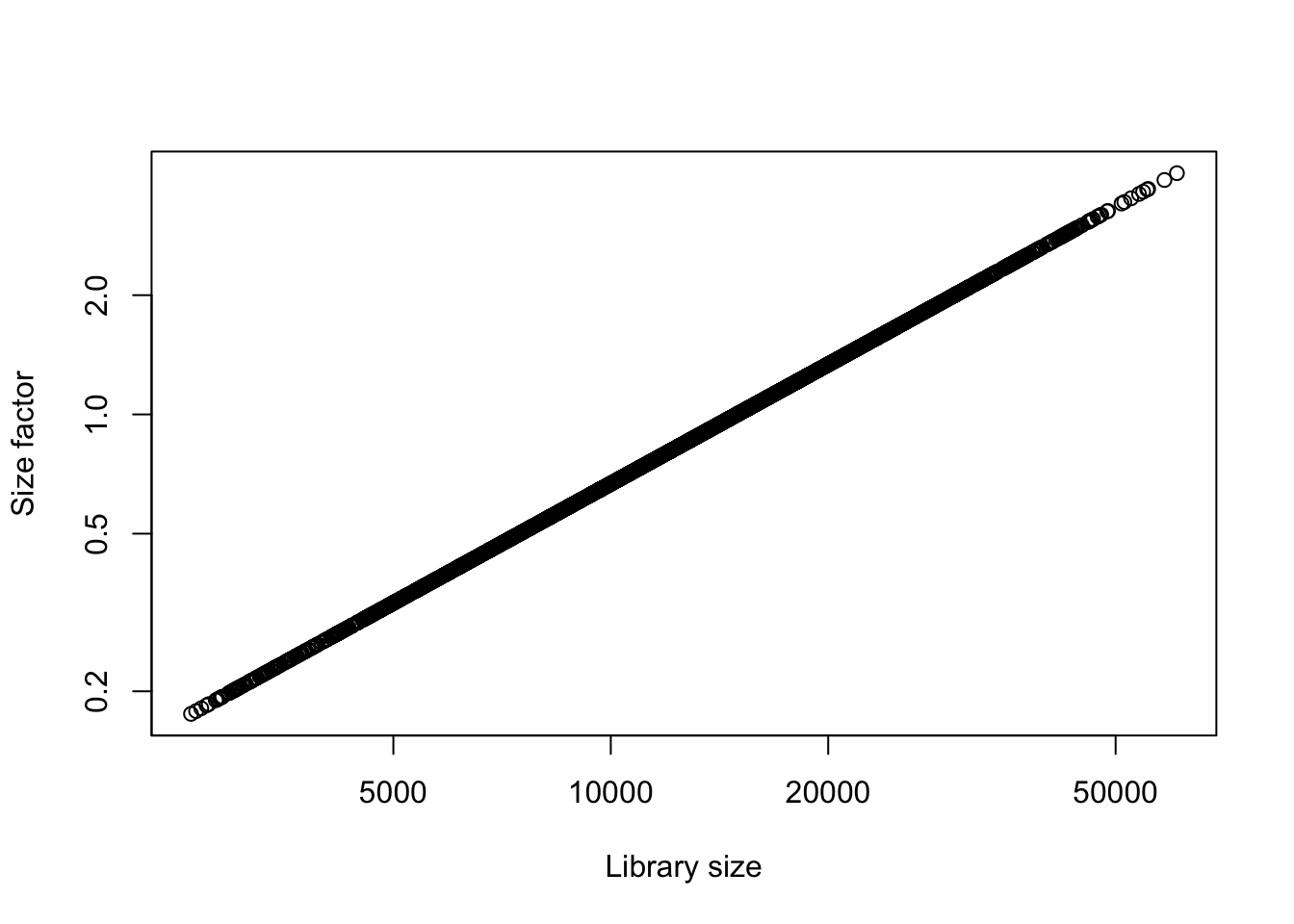
3.4.2 Puntos finales
La normalización por tamaño de biblioteca asume que no hay un sesgo de composición, es decir, si existe un grupo de genes cuya expresión aumenta entonces debe existir otro grupo de genes para los que la expresión disminuye en la misma magnitud, esto generalmente no es verdad para los experimentos de scRNA-seq.
Para algunos análisis exploratorios, la precisión de la normalización no es un punto mayor a considerar. La normalizacion por tamaño de biblioteca suele ser suficiente en algunas ocasiones donde se busca identificar clusters y los marcadores de los clusters, dado que el sesgo por composición normalmente no afecta la separación de los clusters, sólo la magnitud.
Sin embargo, esta falta de precisión podría afectar cuando se busca obtener estimaciones y estadísticas a nivel de genes individuales.
3.5 Normalización por deconvolución (deconvolution)
El problema causado por el sesgo de composición y cómo eliminarlo ha sido estudiado en bulk RNA-seq por métodos como DESeq2::estimateSizeFactorsFromMatrix() y edgeR::calcNormFactors(). En ambos casos, se asume que la mayoría de genes no estarán diferencialmente expresados entre las muestras (en nuestro caso células) y cualquier diferencia entre los genes no diferencialmente expresados representa un sesgo el cual se remueve utilzando el factor de normalización calculado.
Sin embargo, en single-cell RNA-seq se tienen muchas cuentas bajas y ceros debido a limitaciones en la tecnología, las cuales no necesariamente indican ausencia de expresión. Para este fenómento no están preparados los métodos utilizados en bulk RNA-seq.
La normalización por deconvolución (o normalización scran):
- Junta las cuentas de varias células (pool) para incrementar el tamaño de las cuentas, reduciendo ceros y cuentas bajas.
- Aplica los métodos de normalización que corrigen el sesgo de composición.
- Repiten esto para conjuntos sobrelapantes de células para obtener size factors en pool.
- Deconvoluciona estos size factors en pool para obtener size factors para cada célula.
# Normalización por deconvolución (deconvolution)
library("scran")
# Pre-clustering (establece una semilla para obtener resultados reproducibles)
set.seed(100)
clust.zeisel <- quickCluster(sce.zeisel)
# Calcula factores de tamaño para la deconvolución (deconvolution)
deconv.sf.zeisel <-
calculateSumFactors(sce.zeisel, clusters = clust.zeisel, min.mean = 0.1)El pre-clustering mejora la estimación de los size factors al normalizar células similares juntas.
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.1282 0.4859 0.8248 1.0000 1.3194 4.6521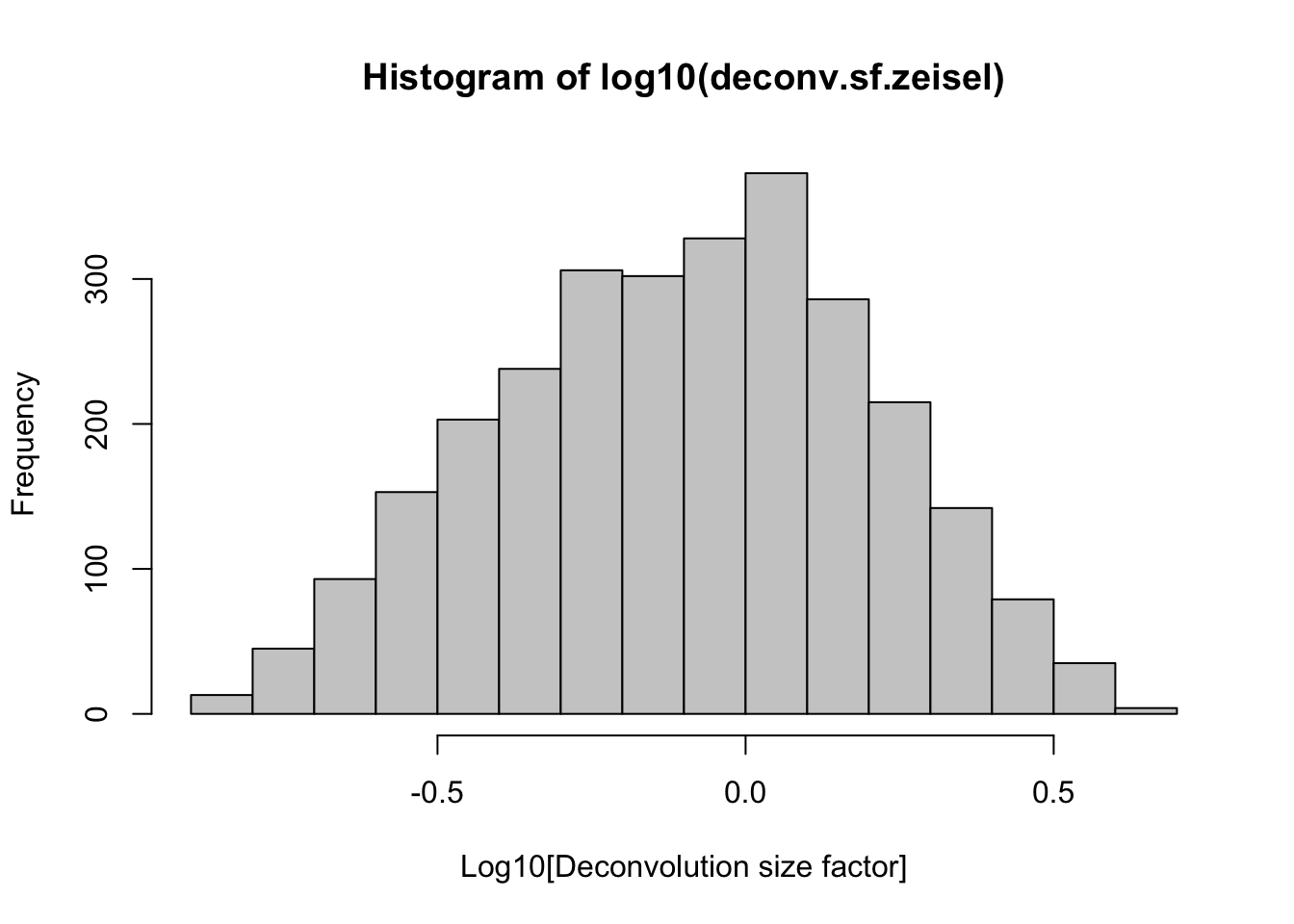
plot(lib.sf.zeisel,
deconv.sf.zeisel,
xlab = "Library size factor",
ylab = "Deconvolution size factor",
log = "xy",
pch = 16,
col=as.integer(factor(sce.zeisel$level1class))
)
abline(a = 0, b = 1, col = "red")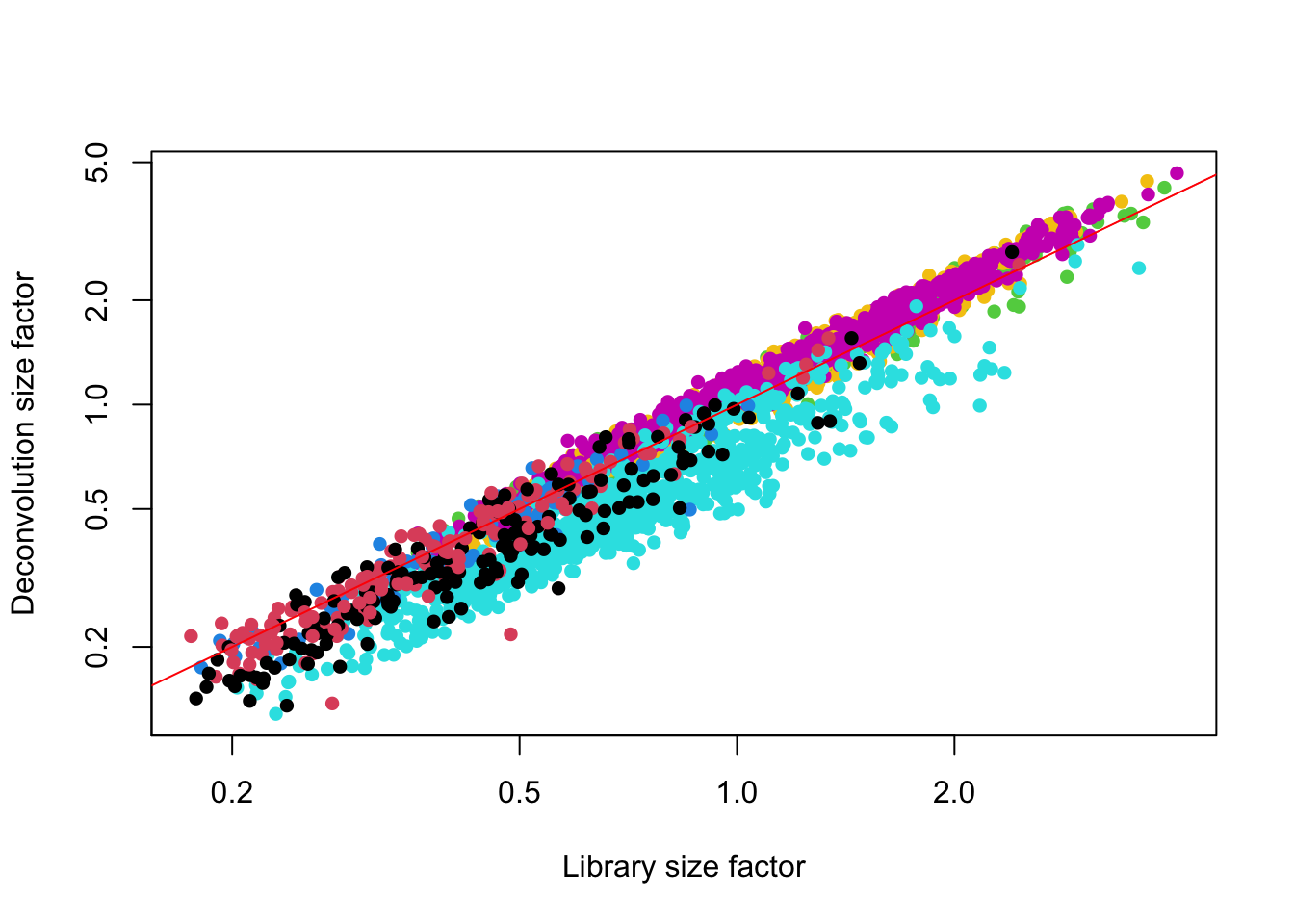
3.5.1 Puntos finales
La normalización por deconvolución (deconvolution) mejora los resultados para análisis posteriores de una manera más precisa que los métodos para bulk RNA-seq.
El método scran::calculateSumFactors algunas veces calcula factores negativos o ceros lo cual altera la matriz de expresión normalizada. ¡Checa los factores que calculas! Si obtienes factores negativos intenta variar el número de clusters.
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.1282 0.4859 0.8248 1.0000 1.3194 4.65213.6 Transformación logarítmica
Una vez calculados los factores de normalización con computeSumFactors(), podemos calcular las cuentas en escala logarítmica usando logNormCounts(). ¿Qué es lo que hace esta función?
- Divide las cuentas de cada gene por el size factor correspondiente para esa célula (estos serían los valores normalizados) y entonces le aplica una transformción logarítmica a estos valores normalizados.
- Típicamente agrega una pseudo-cuenta de 1 para evitar calcular log(0).
- Estas cuentas en escala log se conocen como: valores de expresión normalizados en escala logarítmica (log-transformed normalized expression values).
## class: SingleCellExperiment
## dim: 18441 2815
## metadata(0):
## assays(1): counts
## rownames(18441): ENSMUSG00000029669 ENSMUSG00000046982 ... ENSMUSG00000064337 ENSMUSG00000065947
## rowData names(2): featureType originalName
## colnames(2815): 1772071015_C02 1772071017_G12 ... 1772063068_D01 1772066098_A12
## colData names(10): tissue group # ... level1class level2class
## reducedDimNames(0):
## mainExpName: endogenous
## altExpNames(2): ERCC repeat# Normalization
# Esto agrega el "sizeFactor" como parte del objeto
# set.seed(100)
# clust.zeisel <- quickCluster(sce.zeisel)
sce.zeisel <- computeSumFactors(sce.zeisel, cluster=clust.zeisel, min.mean=0.1)
sce.zeisel## class: SingleCellExperiment
## dim: 18441 2815
## metadata(0):
## assays(1): counts
## rownames(18441): ENSMUSG00000029669 ENSMUSG00000046982 ... ENSMUSG00000064337 ENSMUSG00000065947
## rowData names(2): featureType originalName
## colnames(2815): 1772071015_C02 1772071017_G12 ... 1772063068_D01 1772066098_A12
## colData names(11): tissue group # ... level2class sizeFactor
## reducedDimNames(0):
## mainExpName: endogenous
## altExpNames(2): ERCC repeat## [1] "counts" "logcounts"3.7 Otras normalizaciones
Te invitamos a leer más sobre otras formas de normalizar, para empezar puedes consultar el siguiente curso del Sanger Institute.
Si estas interesad@ en diferencias en el contenido total de RNA en cada célula, checa la normalización por spike-ins. La cual asume que los spike-ins fueron añadidos en un nivel constante en cada célula.
Si tienes resultados donde el library size está asociado a tus datos a pesar de haber normalizado, checa la opción de downsample=TRUE dentro de la función de logNormCounts().
Si te gustaría tener factores de normalización específicos para cada gen que tomen en cuenta la varianza, checa las funciones varianceStabilizingTransformation() de DESeq2 o sctransform de Seurat.
3.10 Agradecimientos
Este curso está basado en el libro Orchestrating Single Cell Analysis with Bioconductor de Aaron Lun, Robert Amezquita, Stephanie Hicks y Raphael Gottardo, además del curso de scRNA-seq para WEHI creado por Peter Hickey.
Y en el material de la comunidadbioinfo/cdsb2020 con el permiso de Leonardo Collado-Torres.
3.11 Detalles de la sesión de R
## [1] "2023-08-10 10:09:19 EDT"## user system elapsed
## 1770.128 124.672 136392.083## ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
## setting value
## version R version 4.3.1 (2023-06-16)
## os macOS Ventura 13.4.1
## system aarch64, darwin20
## ui RStudio
## language (EN)
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz America/New_York
## date 2023-08-10
## rstudio 2023.06.0+421 Mountain Hydrangea (desktop)
## pandoc 3.1.1 @ /Applications/RStudio.app/Contents/Resources/app/quarto/bin/tools/ (via rmarkdown)
##
## ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
## package * version date (UTC) lib source
## abind 1.4-5 2016-07-21 [1] CRAN (R 4.3.0)
## AnnotationDbi * 1.62.2 2023-07-02 [1] Bioconductor
## AnnotationFilter * 1.24.0 2023-05-08 [1] Bioconductor
## AnnotationHub * 3.8.0 2023-05-08 [1] Bioconductor
## beachmat 2.16.0 2023-05-08 [1] Bioconductor
## beeswarm 0.4.0 2021-06-01 [1] CRAN (R 4.3.0)
## Biobase * 2.60.0 2023-05-08 [1] Bioconductor
## BiocFileCache * 2.8.0 2023-05-08 [1] Bioconductor
## BiocGenerics * 0.46.0 2023-06-04 [1] Bioconductor
## BiocIO 1.10.0 2023-05-08 [1] Bioconductor
## BiocManager 1.30.21.1 2023-07-18 [1] CRAN (R 4.3.0)
## BiocNeighbors 1.18.0 2023-05-08 [1] Bioconductor
## BiocParallel 1.34.2 2023-05-28 [1] Bioconductor
## BiocSingular 1.16.0 2023-05-08 [1] Bioconductor
## BiocVersion 3.17.1 2022-12-20 [1] Bioconductor
## biomaRt 2.56.1 2023-06-11 [1] Bioconductor
## Biostrings 2.68.1 2023-05-21 [1] Bioconductor
## bit 4.0.5 2022-11-15 [1] CRAN (R 4.3.0)
## bit64 4.0.5 2020-08-30 [1] CRAN (R 4.3.0)
## bitops 1.0-7 2021-04-24 [1] CRAN (R 4.3.0)
## blob 1.2.4 2023-03-17 [1] CRAN (R 4.3.0)
## bluster * 1.10.0 2023-05-08 [1] Bioconductor
## bookdown 0.34 2023-05-09 [1] CRAN (R 4.3.0)
## bslib 0.5.0 2023-06-09 [1] CRAN (R 4.3.0)
## cachem 1.0.8 2023-05-01 [1] CRAN (R 4.3.0)
## cli 3.6.1 2023-03-23 [1] CRAN (R 4.3.0)
## cluster 2.1.4 2022-08-22 [1] CRAN (R 4.3.1)
## codetools 0.2-19 2023-02-01 [1] CRAN (R 4.3.1)
## colorspace 2.1-0 2023-01-23 [1] CRAN (R 4.3.0)
## cowplot 1.1.1 2020-12-30 [1] CRAN (R 4.3.0)
## crayon 1.5.2 2022-09-29 [1] CRAN (R 4.3.0)
## curl 5.0.1 2023-06-07 [1] CRAN (R 4.3.0)
## DBI 1.1.3 2022-06-18 [1] CRAN (R 4.3.0)
## dbplyr * 2.3.3 2023-07-07 [1] CRAN (R 4.3.0)
## DelayedArray 0.26.7 2023-07-28 [1] Bioconductor
## DelayedMatrixStats 1.22.1 2023-06-09 [1] Bioconductor
## digest 0.6.33 2023-07-07 [1] CRAN (R 4.3.0)
## dplyr * 1.1.2 2023-04-20 [1] CRAN (R 4.3.0)
## dqrng 0.3.0 2021-05-01 [1] CRAN (R 4.3.0)
## DropletUtils * 1.20.0 2023-05-08 [1] Bioconductor
## edgeR 3.42.4 2023-06-04 [1] Bioconductor
## ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.3.0)
## EnsDb.Hsapiens.v86 * 2.99.0 2023-07-29 [1] Bioconductor
## ensembldb * 2.24.0 2023-05-08 [1] Bioconductor
## evaluate 0.21 2023-05-05 [1] CRAN (R 4.3.0)
## ExperimentHub 2.8.1 2023-07-16 [1] Bioconductor
## fansi 1.0.4 2023-01-22 [1] CRAN (R 4.3.0)
## farver 2.1.1 2022-07-06 [1] CRAN (R 4.3.0)
## fastmap 1.1.1 2023-02-24 [1] CRAN (R 4.3.0)
## filelock 1.0.2 2018-10-05 [1] CRAN (R 4.3.0)
## FNN 1.1.3.2 2023-03-20 [1] CRAN (R 4.3.0)
## generics 0.1.3 2022-07-05 [1] CRAN (R 4.3.0)
## GenomeInfoDb * 1.36.1 2023-07-02 [1] Bioconductor
## GenomeInfoDbData 1.2.10 2023-06-08 [1] Bioconductor
## GenomicAlignments 1.36.0 2023-05-08 [1] Bioconductor
## GenomicFeatures * 1.52.1 2023-07-02 [1] Bioconductor
## GenomicRanges * 1.52.0 2023-05-08 [1] Bioconductor
## ggbeeswarm 0.7.2 2023-04-29 [1] CRAN (R 4.3.0)
## ggplot2 * 3.4.2 2023-04-03 [1] CRAN (R 4.3.0)
## ggrepel * 0.9.3 2023-02-03 [1] CRAN (R 4.3.0)
## glue 1.6.2 2022-02-24 [1] CRAN (R 4.3.0)
## gridExtra 2.3 2017-09-09 [1] CRAN (R 4.3.0)
## gtable 0.3.3 2023-03-21 [1] CRAN (R 4.3.0)
## HDF5Array 1.28.1 2023-05-08 [1] Bioconductor
## here 1.0.1 2020-12-13 [1] CRAN (R 4.3.0)
## highr 0.10 2022-12-22 [1] CRAN (R 4.3.0)
## hms 1.1.3 2023-03-21 [1] CRAN (R 4.3.0)
## htmltools 0.5.5 2023-03-23 [1] CRAN (R 4.3.0)
## httpuv 1.6.11 2023-05-11 [1] CRAN (R 4.3.0)
## httr 1.4.6 2023-05-08 [1] CRAN (R 4.3.0)
## igraph 1.5.0.1 2023-07-23 [1] CRAN (R 4.3.0)
## interactiveDisplayBase 1.38.0 2023-05-08 [1] Bioconductor
## IRanges * 2.34.1 2023-07-02 [1] Bioconductor
## irlba 2.3.5.1 2022-10-03 [1] CRAN (R 4.3.0)
## jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.3.0)
## jsonlite 1.8.7 2023-06-29 [1] CRAN (R 4.3.0)
## kableExtra * 1.3.4 2021-02-20 [1] CRAN (R 4.3.0)
## KEGGREST 1.40.0 2023-05-08 [1] Bioconductor
## knitr 1.43 2023-05-25 [1] CRAN (R 4.3.0)
## labeling 0.4.2 2020-10-20 [1] CRAN (R 4.3.0)
## later 1.3.1 2023-05-02 [1] CRAN (R 4.3.0)
## lattice 0.21-8 2023-04-05 [1] CRAN (R 4.3.1)
## lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.3.0)
## lifecycle 1.0.3 2022-10-07 [1] CRAN (R 4.3.0)
## limma 3.56.2 2023-06-04 [1] Bioconductor
## locfit 1.5-9.8 2023-06-11 [1] CRAN (R 4.3.0)
## magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.3.0)
## Matrix * 1.6-0 2023-07-08 [1] CRAN (R 4.3.0)
## MatrixGenerics * 1.12.3 2023-07-30 [1] Bioconductor
## matrixStats * 1.0.0 2023-06-02 [1] CRAN (R 4.3.0)
## memoise 2.0.1 2021-11-26 [1] CRAN (R 4.3.0)
## metapod 1.8.0 2023-04-25 [1] Bioconductor
## mime 0.12 2021-09-28 [1] CRAN (R 4.3.0)
## munsell 0.5.0 2018-06-12 [1] CRAN (R 4.3.0)
## patchwork * 1.1.2 2022-08-19 [1] CRAN (R 4.3.0)
## PCAtools * 2.12.0 2023-05-08 [1] Bioconductor
## pheatmap * 1.0.12 2019-01-04 [1] CRAN (R 4.3.0)
## pillar 1.9.0 2023-03-22 [1] CRAN (R 4.3.0)
## pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.3.0)
## plyr 1.8.8 2022-11-11 [1] CRAN (R 4.3.0)
## png 0.1-8 2022-11-29 [1] CRAN (R 4.3.0)
## prettyunits 1.1.1 2020-01-24 [1] CRAN (R 4.3.0)
## progress 1.2.2 2019-05-16 [1] CRAN (R 4.3.0)
## promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.3.0)
## ProtGenerics 1.32.0 2023-05-08 [1] Bioconductor
## purrr 1.0.1 2023-01-10 [1] CRAN (R 4.3.0)
## R.methodsS3 1.8.2 2022-06-13 [1] CRAN (R 4.3.0)
## R.oo 1.25.0 2022-06-12 [1] CRAN (R 4.3.0)
## R.utils 2.12.2 2022-11-11 [1] CRAN (R 4.3.0)
## R6 2.5.1 2021-08-19 [1] CRAN (R 4.3.0)
## rappdirs 0.3.3 2021-01-31 [1] CRAN (R 4.3.0)
## RColorBrewer 1.1-3 2022-04-03 [1] CRAN (R 4.3.0)
## Rcpp 1.0.11 2023-07-06 [1] CRAN (R 4.3.0)
## RCurl 1.98-1.12 2023-03-27 [1] CRAN (R 4.3.0)
## reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.3.0)
## restfulr 0.0.15 2022-06-16 [1] CRAN (R 4.3.0)
## rhdf5 2.44.0 2023-05-08 [1] Bioconductor
## rhdf5filters 1.12.1 2023-05-08 [1] Bioconductor
## Rhdf5lib 1.22.0 2023-05-08 [1] Bioconductor
## rjson 0.2.21 2022-01-09 [1] CRAN (R 4.3.0)
## rlang 1.1.1 2023-04-28 [1] CRAN (R 4.3.0)
## rmarkdown 2.23 2023-07-01 [1] CRAN (R 4.3.0)
## rprojroot 2.0.3 2022-04-02 [1] CRAN (R 4.3.0)
## Rsamtools 2.16.0 2023-06-04 [1] Bioconductor
## RSQLite 2.3.1 2023-04-03 [1] CRAN (R 4.3.0)
## rstudioapi 0.15.0 2023-07-07 [1] CRAN (R 4.3.0)
## rsvd 1.0.5 2021-04-16 [1] CRAN (R 4.3.0)
## rtracklayer 1.60.0 2023-05-08 [1] Bioconductor
## Rtsne 0.16 2022-04-17 [1] CRAN (R 4.3.0)
## rvest 1.0.3 2022-08-19 [1] CRAN (R 4.3.0)
## S4Arrays 1.0.5 2023-07-24 [1] Bioconductor
## S4Vectors * 0.38.1 2023-05-08 [1] Bioconductor
## sass 0.4.7 2023-07-15 [1] CRAN (R 4.3.0)
## ScaledMatrix 1.8.1 2023-05-08 [1] Bioconductor
## scales 1.2.1 2022-08-20 [1] CRAN (R 4.3.0)
## scater * 1.28.0 2023-04-25 [1] Bioconductor
## scran * 1.28.2 2023-07-23 [1] Bioconductor
## scRNAseq * 2.14.0 2023-04-27 [1] Bioconductor
## scuttle * 1.9.4 2023-01-23 [1] Bioconductor
## sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.3.0)
## shiny 1.7.4.1 2023-07-06 [1] CRAN (R 4.3.0)
## SingleCellExperiment * 1.22.0 2023-05-08 [1] Bioconductor
## sparseMatrixStats 1.12.2 2023-07-02 [1] Bioconductor
## statmod 1.5.0 2023-01-06 [1] CRAN (R 4.3.0)
## stringi 1.7.12 2023-01-11 [1] CRAN (R 4.3.0)
## stringr 1.5.0 2022-12-02 [1] CRAN (R 4.3.0)
## SummarizedExperiment * 1.30.2 2023-06-06 [1] Bioconductor
## svglite 2.1.1 2023-01-10 [1] CRAN (R 4.3.0)
## systemfonts 1.0.4 2022-02-11 [1] CRAN (R 4.3.0)
## tibble 3.2.1 2023-03-20 [1] CRAN (R 4.3.0)
## tidyselect 1.2.0 2022-10-10 [1] CRAN (R 4.3.0)
## utf8 1.2.3 2023-01-31 [1] CRAN (R 4.3.0)
## uwot 0.1.16 2023-06-29 [1] CRAN (R 4.3.0)
## vctrs 0.6.3 2023-06-14 [1] CRAN (R 4.3.0)
## vipor 0.4.5 2017-03-22 [1] CRAN (R 4.3.0)
## viridis 0.6.4 2023-07-22 [1] CRAN (R 4.3.0)
## viridisLite 0.4.2 2023-05-02 [1] CRAN (R 4.3.0)
## webshot 0.5.5 2023-06-26 [1] CRAN (R 4.3.0)
## withr 2.5.0 2022-03-03 [1] CRAN (R 4.3.0)
## xfun 0.39 2023-04-20 [1] CRAN (R 4.3.0)
## XML 3.99-0.14 2023-03-19 [1] CRAN (R 4.3.0)
## xml2 1.3.5 2023-07-06 [1] CRAN (R 4.3.0)
## xtable 1.8-4 2019-04-21 [1] CRAN (R 4.3.0)
## XVector 0.40.0 2023-05-08 [1] Bioconductor
## yaml 2.3.7 2023-01-23 [1] CRAN (R 4.3.0)
## zlibbioc 1.46.0 2023-05-08 [1] Bioconductor
##
## [1] /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/library
##
## ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────