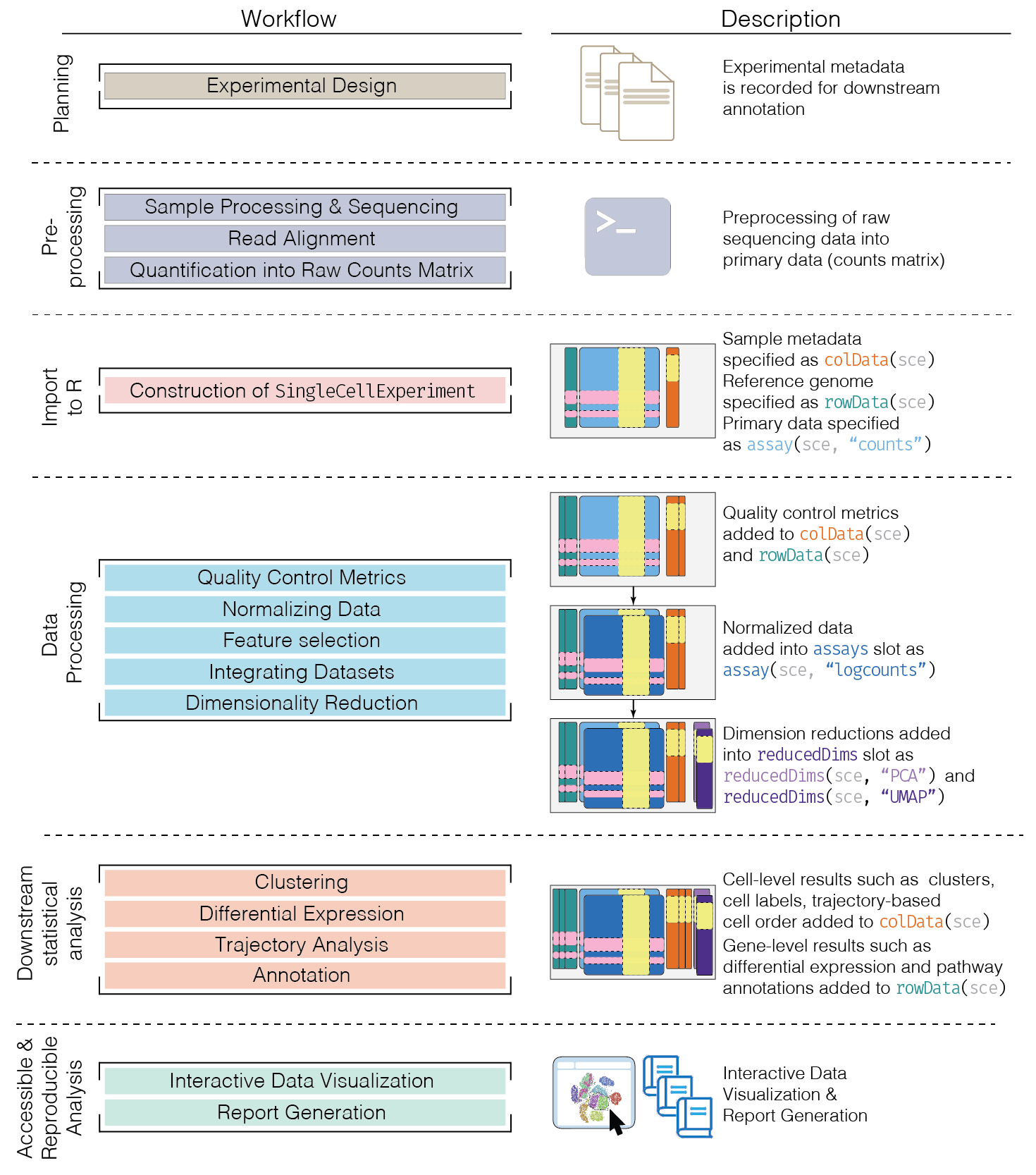
1 Estructura e importe de datos
Joselyn Cristina Chávez Fuentes
07 de agosto de 2023
1.2 Bulk RNAseq vs single-cell RNAseq
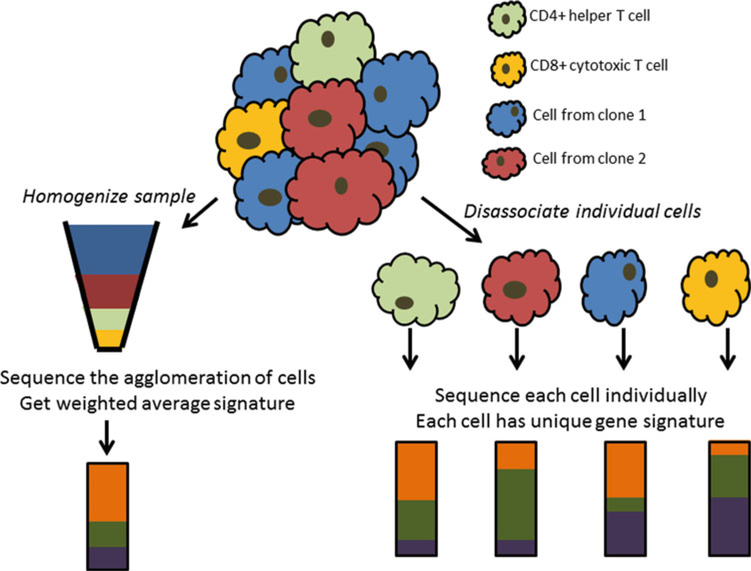 Tomado de Yu X, et al.
Tomado de Yu X, et al.
- Bulk RNAseq
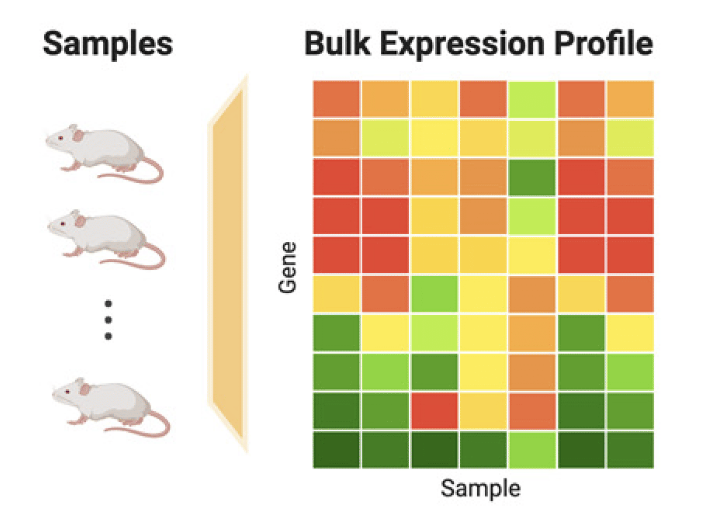 Tomado de Hyeongseon Jeon, et al.
Tomado de Hyeongseon Jeon, et al.
- scRNAseq
1.3 Consideraciones experimentales
1.3.1 Tecnologías de scRNAseq
- Droplet-based:
Son las tecnologías más usadas debido a su buen rendimiento con un relativo bajo costo.
10X Genomics
inDrop seq
Drop-seq
Plate-based
Tienen la capacidad de capturar información adicional, como la morfología. Tienen más opciones para personalizar el diseño de experimento.
- Con UMIs:
- CEL-seq
- MARS-seq
- Con reads:
- Smart-seq2
- Otros
- sciRNA-seq
1.3.2 ¿UMIs o Reads?
Los métodos basados en reads proveen una cobertura de transcriptoma completo, lo que puede ser útil para ciertas aplicaciones, como el análisis de splicing o mutaciones en exones.
Los métodos basados en UMIs suelen ser más populares, ya que eliminan el ruido causado por la amplificación durante el PCR.
1.3.3 ¿Cuántas células y profundidad de secuenciación necesito?
Depende…
Si tu objetivo es el estudio de subgrupos raros o poco abundantes de células, necesitarás un mayor número de células por muestra.
Si tu objetivo es estudiar diferencias sutiles en la expresión de genes, necesitarás una mayor profundidad de secuenciación.
Hasta el momento, las tecnologías basadas en droplets capturan entre 10,000 y 100,000 células, con un aproximado de 1,000 a 10,000 UMIs por célula.
Existen algunas variaciones entre el rendimiento y la tasa de doublets que pueden afectar la eficiencia real de secuenciación.
Encontrar un balance entre el número de células por muestra, la profundidad de secuenciación, el número de condiciones y réplicas a secuenciar depende mucho de la aplicación y el presupuesto.
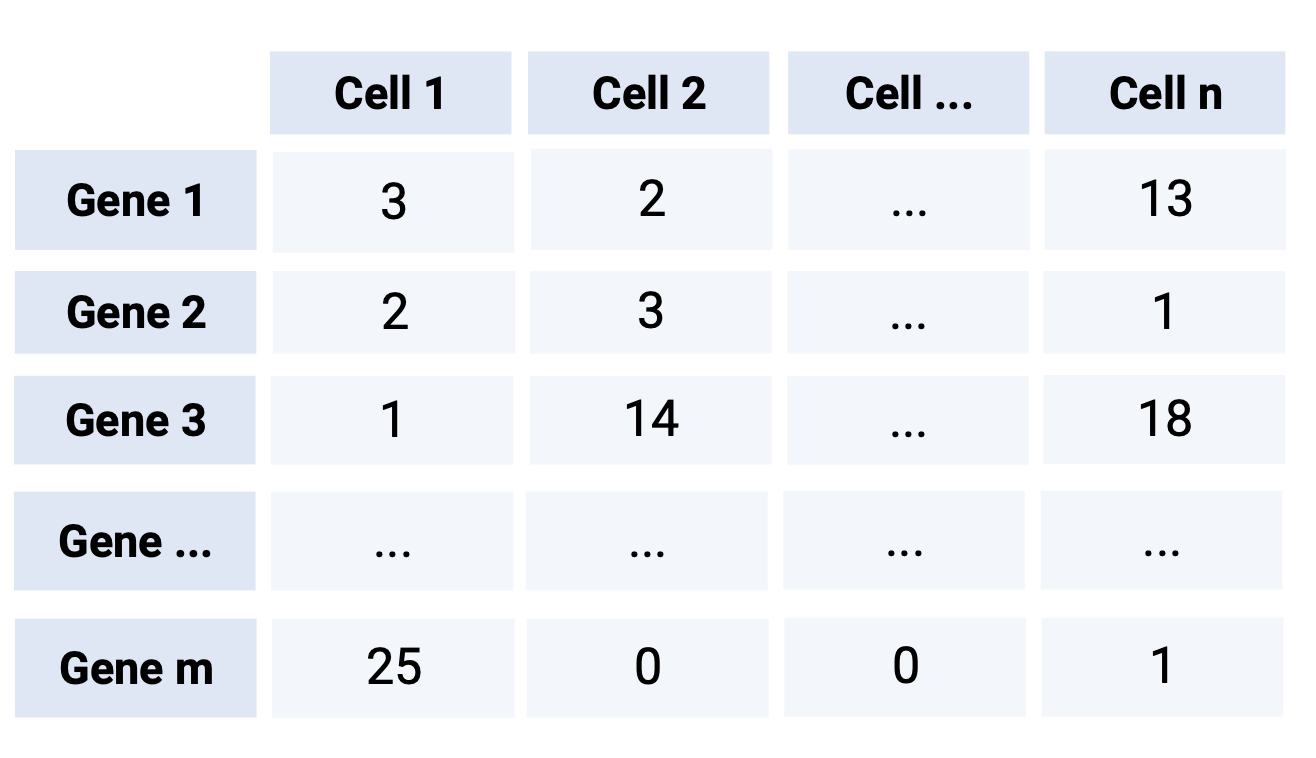
1.4 Generación de la matriz de cuentas
El método para generar la matriz depende de la tecnología utilizada.
10X Genomics:
Cellranger es el programa más popular. Utiliza STAR para alinear los reads con el genoma de referencia y después cuenta el número de UMIs únicos mapeados con cada gene.
Como alternativa, métodos de pseudo-alineamiento como alevin pueden ser usados. No requiere un alineamiento con genoma de referencia, lo que reduce el tiempo de cómputo y los requerimientos de memoria.
El paquete scPipe provee un análisis general. Utiliza Rsubread para alinear los reads y después cuenta reads o UMIs por gene.
CEL-seq o CEL-seq2
- El paquete scruff provee un pipeline para la cuantificación.
De manera general, los protocolos basados en reads pueden reusar los métodos desarrollados para bulk RNA-seq.
Si el grupo de datos involucra transcritos spike-in, las secuencias spike-in deben proveerse junto al genoma de referencia durante el alineamiento y cuantificación.
1.5 Nombres de los genes
En todos los casos, los identificadores de genes deben definirse considerando los nombres de Ensembl o Entrez. Esto mantiene un mapeo sin lugar a errores para la identificación de genes en la matriz.
Estos identificadores pueden ser reemplazados por el nombre común del gene durante el análisis, pero considera que estos nombres pueden cambiar con el tiempo.
Consideraciones adicionales:
Algunas herramientas de conteo, como HTSeq, incluyen un reporte dentro de la matriz de cuentas con el número lecturas sin alinear. Estos valores pueden ser útiles para el control de calidad, pero deben ser removidas de la matriz de cuentas y guardadas en otro lugar antes de continuar con el análisis para que no sean confundidas con los valores de expresión de genes.
Generalmente, las secuencias spike-ins son desarrolladas por el External RNA Controls Consortium (ERCC) y suelen tener nombres como ERCC-00002. En las muestra de humano, debemos evitar confundir estos identificadores con los genes de la familia ERCC, que suelen tener nombres similares ERCC1. Podemos evitar estos problemas si usamos los identificadores de Ensembl.
1.6 Importando los datos
1.6.1 Datos tabulares
Descarguemos los datos de cáncer de páncreas de Muraro et al (2016) GSE85241.
Solamente necesitamos el archivo “GSE85241_cellsystems_dataset_4donors_updated.csv”
- Una forma sencilla
mat <- read.delim(
"GSE85241_cellsystems_dataset_4donors_updated.csv")
mat <- as.matrix(mat)
dim(mat)
mat[1:3,1:3]- Una opción más eficiente. Solamente se leen los datos diferentes a cero.
1.7 Actividad
Comparemos la clase y la cantidad de memoria que utiliza cada opción.
Utiliza las funciones class() y lobstr::obj_size() para comparar mat y sparse.mat
1.7.1 Datos de cellRanger
Cuando usamos datos de 10X Genomics, Cellranger genera un directorio que contiene 3 archivos: cuentas, anotaciones de features y barcodes. Podemos usar la ruta a este directorio y la función read10xCounts() del paquete DropletUtils.
Utilicemos como ejemplo un dataset de células sanguíneas periféricas.
1.8 Actividad
Evalúa la clase de sce. ¿Habías escuchado antes sobre este tipo de objeto?
Imprime el objeto sce en la consola. ¿Cómo se ve el resultado?
1.8.1 Datos con formato HDF5
El formato Hierarchical Data Format version 5 (HDF5) permite guardar tanto los valores de expresión asociados a los genes, como la anotación de los tipos celulares dentro de un mismo archivo.
Podemos leer este archivo con un objeto SingleCellExperiment utilizando el paquete zellkonverter.
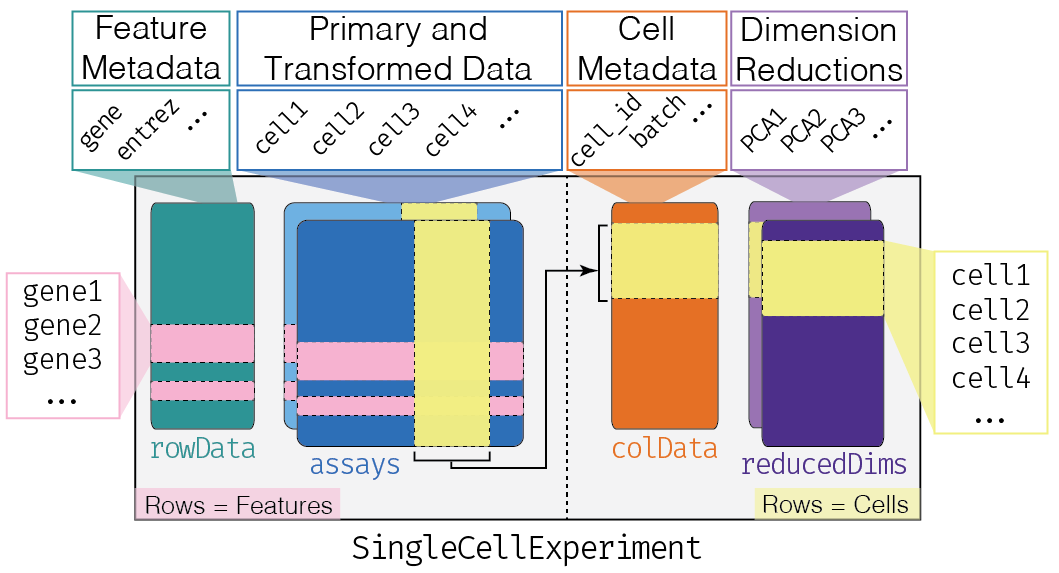
1.10 Construyendo un objeto SingleCellExperiment
Para crear un SingleCellExperiment rudimentario, solamente necesitamos el slot assays.
Este slot necesita la matriz de cuentas, donde los renglones corresponden a los genes (features) y las columnas corresponden a las células.
1.11 Accediendo a los elementos del objeto
Para poder ver el contenido del slot assays tenemos dos opciones:
- Forma general, usando el nombre del assay que queremos ver:
- Una opción más corta, pero sólo funciona cuando el assay se llama “counts”