2 Exploración interactiva de datos transcriptómicos con iSEE
El código de R de este archivo está disponbie vía GitHub.
## Carguemos las librerías que vamos a usar para esta parte del mini curso
## Infraestructura
library("SummarizedExperiment")## Loading required package: MatrixGenerics## Loading required package: matrixStats##
## Attaching package: 'matrixStats'## The following object is masked from 'package:dplyr':
##
## count##
## Attaching package: 'MatrixGenerics'## The following objects are masked from 'package:matrixStats':
##
## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse, colCounts, colCummaxs, colCummins,
## colCumprods, colCumsums, colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs, colMads, colMaxs,
## colMeans2, colMedians, colMins, colOrderStats, colProds, colQuantiles, colRanges, colRanks, colSdDiffs,
## colSds, colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads, colWeightedMeans,
## colWeightedMedians, colWeightedSds, colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods, rowCumsums, rowDiffs, rowIQRDiffs,
## rowIQRs, rowLogSumExps, rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins, rowOrderStats,
## rowProds, rowQuantiles, rowRanges, rowRanks, rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs,
## rowVars, rowWeightedMads, rowWeightedMeans, rowWeightedMedians, rowWeightedSds, rowWeightedVars## Loading required package: GenomicRanges## Loading required package: stats4## Loading required package: BiocGenerics## Loading required package: parallel##
## Attaching package: 'BiocGenerics'## The following objects are masked from 'package:parallel':
##
## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ, clusterExport, clusterMap, parApply,
## parCapply, parLapply, parLapplyLB, parRapply, parSapply, parSapplyLB## The following objects are masked from 'package:dplyr':
##
## combine, intersect, setdiff, union## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, basename, cbind, colnames, dirname, do.call, duplicated, eval,
## evalq, Filter, Find, get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget, order,
## paste, pmax, pmax.int, pmin, pmin.int, Position, rank, rbind, Reduce, rownames, sapply, setdiff, sort,
## table, tapply, union, unique, unsplit, which.max, which.min## Loading required package: S4Vectors##
## Attaching package: 'S4Vectors'## The following object is masked from 'package:plotly':
##
## rename## The following objects are masked from 'package:dplyr':
##
## first, rename## The following object is masked from 'package:tidyr':
##
## expand## The following object is masked from 'package:base':
##
## expand.grid## Loading required package: IRanges##
## Attaching package: 'IRanges'## The following object is masked from 'package:plotly':
##
## slice## The following objects are masked from 'package:dplyr':
##
## collapse, desc, slice## The following object is masked from 'package:purrr':
##
## reduce## Loading required package: GenomeInfoDb## Loading required package: Biobase## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.##
## Attaching package: 'Biobase'## The following object is masked from 'package:MatrixGenerics':
##
## rowMedians## The following objects are masked from 'package:matrixStats':
##
## anyMissing, rowMedianslibrary("SingleCellExperiment")
## library("SpatialExperiment") ## A partir de Bioconductor 3.13
## que estará disponible a finales de mayo 2021
## Para descargar y procesar datos
library("scRNAseq")
library("scater")
library("Rtsne")
library("recount3")
## Para visualizar los datos
library("iSEE")2.1 Infraestructura para datos tránscriptomicos

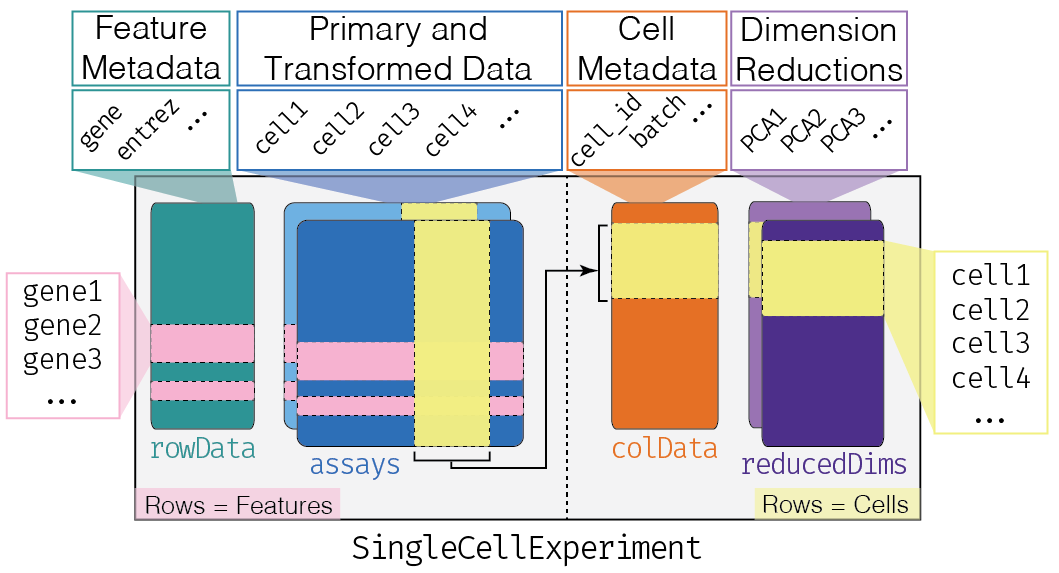
- Revisemos las diapositivas 31 a 43 de la siguiente presentación
Thanks to Pete @PeteHaitch & #OSCA ❤️ I'm now ready for my Human Cell Atlas - Latin America presentation on “Getting started with #scRNA-seq analyses with @Bioconductor” 🎉
— 🇲🇽 Leonardo Collado-Torres (@lcolladotor) May 2, 2021
📔 https://t.co/nEWZcnadyh
💻 https://t.co/7pXDTQRpF0#rstats @CDSBMexico @humancellatlas #LatAm pic.twitter.com/Otg8rdSlYW
- Incluso va más allá con SpatialExperiment
Are you working with spatial transcriptomics data such as Visium from @10xGenomics? Then you'll be interested in #SpatialExperiment 📦 led by @drighelli @lmwebr @CrowellHL with contributions by @PardoBree @shazanfar A Lun @stephaniehicks @drisso1893 🌟
— 🇲🇽 Leonardo Collado-Torres (@lcolladotor) January 29, 2021
📜 https://t.co/r36qlakRJe pic.twitter.com/cWIiwLFitV
- Usamos
SpatialExperimentpara hacer spatialLIBD. En este proyecto participó Brenda Pardo, que es una estudiante de tercer año de la LCG-EJ-UNAM en el LIIGH-UNAM.
Our paper describing our package #spatialLIBD is finally out! 🎉🎉🎉
— Brenda Pardo (@PardoBree) April 30, 2021
spatialLIBD is an #rstats / @Bioconductor package to visualize spatial transcriptomics data.
⁰
This is especially exciting for me as it is my first paper as a first author 🦑.https://t.co/COW013x4GA
1/9 pic.twitter.com/xevIUg3IsA
2.2 Datos públicos
scRNAseq
scRNAseq es un paquete de Bioconductor que creó Aaron Lun para poder tener varios datos de ejemplo de scRNA-seq (secuenciación masiva de ARN de células únicas) para poder demostrar sus métodos estadísticos. Ese paquete lo usan en iSEE para obtener datos de ejemplo y también lo usan mucho en Orchestrating Single Cell Analysis (OSCA) with Bioconductor http://bioconductor.org/books/release/OSCA/.
A continuación vamos a descargar unos datos de scRNA-seq que usan en los ejemplos de iSEE en http://bioconductor.org/packages/release/bioc/vignettes/iSEE/inst/doc/basic.html#2_Setting_up_the_data. scRNAseq usa otro paquete de Bioconductor llamado ExperimentHub para descargar los archivos y organizarlos en un lugar de tal forma que si corremos el código otra vez en la misma computadora, no tendremos que volver a descargar los archivos. Es decir, crea un cache local.
## Descarguemos datos de ejemplo de scRNA-seq del proyecto de
## Tasic et al 2016
sce <- ReprocessedAllenData(assays = "tophat_counts")
sce## class: SingleCellExperiment
## dim: 20816 379
## metadata(2): SuppInfo which_qc
## assays(1): tophat_counts
## rownames(20816): 0610007P14Rik 0610009B22Rik ... Zzef1 Zzz3
## rowData names(0):
## colnames(379): SRR2140028 SRR2140022 ... SRR2139341 SRR2139336
## colData names(22): NREADS NALIGNED ... Animal.ID passes_qc_checks_s
## reducedDimNames(0):
## altExpNames(1): ERCCLos datos que descargamos son datos digamos que crudos, así que necesitamos correr un par de análisis usando scater para que sea más interesante el ejemplo. No es el objetivo de este mini curso explicar esos pasos, aunque a grandes rasgos, nos van a ayudar a que podamos comparar las diferentes células. También nos ayudará a visualizar las células.
## Normalizamos los datos
sce <- logNormCounts(sce, exprs_values = "tophat_counts")
## Obtenemos dimensiones reducidas usando primero un análisis de componentes
## principales y luego con tSNE
set.seed(1000)
sce <- runPCA(sce)
sce <- runTSNE(sce)
## Ahora tenemos datos de PCA y TSNE en nuestro objeto de R
reducedDimNames(sce)## [1] "PCA" "TSNE"## Podemos ahora calcular unas variables a lo largo de nuestros genes
## tal como la expresión media del gene en las células
## y la varianza
rowData(sce)$mean_log <- rowMeans(logcounts(sce))
rowData(sce)$var_log <- apply(logcounts(sce), 1, var)
rowData(sce)## DataFrame with 20816 rows and 2 columns
## mean_log var_log
## <numeric> <numeric>
## 0610007P14Rik 7.9369535 9.37109
## 0610009B22Rik 6.7572139 10.80402
## 0610009L18Rik 0.0881306 0.40976
## 0610009O20Rik 3.6084644 14.97973
## 0610010F05Rik 6.5254121 13.45587
## ... ... ...
## Zyg11a 0.0854769 0.109352
## Zyg11b 8.9813448 3.988757
## Zyx 2.4291810 11.860900
## Zzef1 2.9058253 11.223440
## Zzz3 4.4118629 15.179005Ahora nuestro objeto sce es más complejo y tiene más información que podremos visualizar con iSEE.
recount3
Para que tengan más datos de ejemplo, usaremos datos del proyecto de recount3 vía el cual pueden descargar información de más de 700,000 muestras de RNA-seq de ratón o humano tal como describimos en http://rna.recount.bio/. La idea es que pueden obtener objetos de R del tipo RangedSummarizedExperiment que también podemos visualizar con iSEE.

## Revisemos todos los proyectos con datos de humano en recount3
human_projects <- available_projects()
## Veamos cuantos proyectos hay
nrow(human_projects)## [1] 8742## Encuentra tu proyecto de interés. Aquí usaremos
## SRP009615 de ejemplo
proj_info <- subset(
human_projects,
project == "SRP009615" & project_type == "data_sources"
)
## Crea un objetio de tipo RangedSummarizedExperiment (RSE)
## con la información a nivel de genes
rse_gene_SRP009615 <- create_rse(proj_info)
## Explora el objeto RSE
rse_gene_SRP009615## class: RangedSummarizedExperiment
## dim: 63856 12
## metadata(8): time_created recount3_version ... annotation recount3_url
## assays(1): raw_counts
## rownames(63856): ENSG00000278704.1 ENSG00000277400.1 ... ENSG00000182484.15_PAR_Y ENSG00000227159.8_PAR_Y
## rowData names(10): source type ... havana_gene tag
## colnames(12): SRR387777 SRR387778 ... SRR389077 SRR389078
## colData names(175): rail_id external_id ... recount_pred.curated.cell_line BigWigURLLos objetos de recount3 tienen ciertas propiedades que aquí no explicaremos. Pero bueno, básicamente los siguientes comandos los querrán usar con los diferentes datos que descarguen de recount3.
## Convirtamos las cuentas por nucleotido a cuentas por lectura
## usando compute_read_counts().
## Para otras transformaciones como RPKM y TPM, revisa transform_counts().
assay(rse_gene_SRP009615, "counts") <- compute_read_counts(rse_gene_SRP009615)## Para este estudio en específico, hagamos más fácil de usar la
## información del experimento
rse_gene_SRP009615 <- expand_sra_attributes(rse_gene_SRP009615)
colData(rse_gene_SRP009615)[
,
grepl("^sra_attribute", colnames(colData(rse_gene_SRP009615)))
]## DataFrame with 12 rows and 4 columns
## sra_attribute.cells sra_attribute.shRNA_expression sra_attribute.source_name sra_attribute.treatment
## <character> <character> <character> <character>
## SRR387777 K562 no SL2933 Puromycin
## SRR387778 K562 yes, targeting SRF SL2934 Puromycin, doxycycline
## SRR387779 K562 no SL5265 Puromycin
## SRR387780 K562 yes targeting SRF SL3141 Puromycin, doxycycline
## SRR389079 K562 no shRNA expression SL6485 Puromycin
## ... ... ... ... ...
## SRR389082 K562 expressing shRNA tar.. SL2592 Puromycin, doxycycline
## SRR389083 K562 no shRNA expression SL4337 Puromycin
## SRR389084 K562 expressing shRNA tar.. SL4326 Puromycin, doxycycline
## SRR389077 K562 no shRNA expression SL1584 Puromycin
## SRR389078 K562 expressing shRNA tar.. SL1583 Puromycin, doxycycline2.3 Explorando con iSEE
2.3.1 sce
if (interactive()) {
iSEE(sce, appTitle = "Aprendiendo a usar iSEE")
}- Exploremos varias variables (column data) en los PCA y TSNE. ¿Qué variable separa los grupos de muestras?
- ¿Qué muestra tiene la mayor cantidad de número de lecturas (NREADS)?
- Aprendamos a hacer _facet_s en el panel que muestra información de los genes (feature assay plot 1) y a ligar ese panel a la tabla con información de los genes
- Comparemos el número de lecturas (NREADS) usando _facet_s por disección. ¿Alguna nos llama la atención? Podemos usar la columna de “passes qc checks”.
- Hagamos una gráfica de la media (eje X) vs la varianza (eje Y) de expresión de los genes. ¿Vemos algún patrón?
2.3.2 Ejercicio
if (interactive()) {
iSEE(rse_gene_SRP009615, appTitle = "iSEE con datos de recount3")
}2.3.3 Datos reales
Datos de https://www.biorxiv.org/content/10.1101/2020.10.07.329839v1.full que pueden ver en pagínas web creadas con iSEE a través de https://github.com/LieberInstitute/10xPilot_snRNAseq-human#explore-the-data-interactively. Para lograr esto subimos el resultado de iSEE a https://shinyapps.io.
2.4 Más material
Si les llamó la atención iSEE y recount3, vía https://lcolladotor.github.io/rnaseq_LCG-UNAM_2021 pueden aprender más sobre estos paquetes.
