4 Estructura e importe de datos
Instructoras: Elisa Márquez Zavala, Citlali Gil Aguillon.
Contenido adaptado de CDSB2020: Introducción a scRNA-seq, estructura e importe de datos de Leonardo Collado Torres.
4.1 Preprocesamiento de datos
4.1.1 Cell Ranger
cellrangeres un pipeline que provee 10x Genomics para procesar datos de scRNA-seq generados con la tecnología de Chromium de 10x Genomics
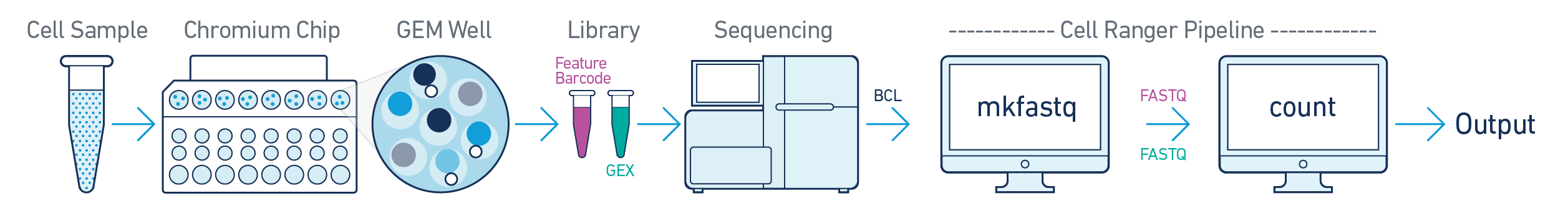
cellranger countprocesa los datos crudos de FASTQ y genera los archivos que podemos leer en R usandoDropletUtils::read10xCounts().
4.1.2 scPipe
scPipees otro pipeline para procesar datos crudos (en formato FASTQ) de tecnologías de scRNA-seq. Funciona para varias tecnologías y usaRsubreadpara el paso de alineamiento.
we developed scPipe, an R/Bioconductor package that integrates barcode demultiplexing, read alignment, UMI-aware gene-level quantification and quality control of raw sequencing data generated by multiple protocols that include CEL-seq, MARS-seq, Chromium 10X, Drop-seq and Smart-seq.

Fuente: Tian et al, PLoS Comput Biol., 2018.
4.1.3 Etc
Hay muchos pipelines para procesar datos de scRNA-seq, porque a diferencia de bulk RNA-seq, en scRNA-seq hay muchas tecnologías en el mercado. Aunque tal vez la más común es la de 10x Genomics Chromium.
4.2 Estructura de SingleCellExperiment
Dentro del curso -y más ampliamente dentro de los análisis de scRNA-seq en R- emplearemos la clase SingleCellExperiment .
Además recordemos que la interoperabilidad dentro de los paquetes de Bioconductor hará que facilmente puedas ajustarte más facilmente a otros paquetes que vayamos encontrando útiles (la infraestructura de los dato nos seguirá sirviendo!).
Podríamos dividir esta clase en cuatro categorias:
datos primarios y transformados (donde estaran lo)
metadata de datos (información de los genes o features, de las células y del experimento)
reduccion de dimensiones
experimentos alternativos
Todo lo anterior está ligado, por lo que hace más sencillo el manejo de subsets de interés
por ejemplo si nos interesa los genes x,y,z podríamos solicitarlos y con ello traer la información de las demás tablas
Examinaremos cada una de estas partes a detalle
Usaremos las diapositivas de Peter Hickey para explicar la clase SingleCellExperiment en las siguientes diapositivas. A la par se demostrará el siguiente código.
4.3 Ejercicio 1
The A. T. L. Lun et al. (2017) dataset contains two 96-well plates of 416B cells (an immortalized mouse myeloid progenitor cell line), processed using the Smart-seq2 protocol (Picelli et al. 2014). A constant amount of spike-in RNA from the External RNA Controls Consortium (ERCC) was also added to each cell’s lysate prior to library preparation. High-throughput sequencing was performed and the expression of each gene was quantified by counting the total number of reads mapped to its exonic regions. Similarly, the quantity of each spike-in transcript was measured by counting the number of reads mapped to the spike-in reference sequences.
Fragmento obtenido de https://bioconductor.org/books/release/OSCA/lun-416b-cell-line-smart-seq2.html
library("scRNAseq")
sce.416b <- LunSpikeInData(which = "416b")
# Carga el paquete SingleCellExperiment
library("SingleCellExperiment")
"Primera parte
aquí checamos el slot assays"## [1] "Primera parte\naquí checamos el slot assays"# Extrae la matriz de cuentas del set de datos de 416b
counts.416b <- counts(sce.416b)
# CHEQUEMOS clase y dimensiones
class(counts.416b) # es matriz## [1] "matrix" "array"dim(counts.416b) # indicará genes y células## [1] 46604 192# CONSTRUIR un nuevo objeto SCE de la matriz de cuentas !!!!!!
sce <- SingleCellExperiment(assays = list(counts = counts.416b))
# Revisa el objeto que acabamos de crear
sce## class: SingleCellExperiment
## dim: 46604 192
## metadata(0):
## assays(1): counts
## rownames(46604): ENSMUSG00000102693 ENSMUSG00000064842 ...
## ENSMUSG00000095742 CBFB-MYH11-mcherry
## rowData names(0):
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(0):
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce) / 1024^2 ## En MB## 38.26031 B# Accesa la matriz de cuenta del compartimento (slot) "assays"
# assays(sce, "counts")
# OJO: ¡esto puede inundar tu sesión de R!
# 1. El método general
assay(sce, "counts")[110:115, 1:3] # gene, cell## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 0
## ENSMUSG00000025915 8
## ENSMUSG00000046334 2
## ENSMUSG00000088916 0
## ENSMUSG00000046101 0
## ENSMUSG00000098234 63
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 0
## ENSMUSG00000025915 0
## ENSMUSG00000046334 3
## ENSMUSG00000088916 0
## ENSMUSG00000046101 0
## ENSMUSG00000098234 21
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 19
## ENSMUSG00000025915 1
## ENSMUSG00000046334 15
## ENSMUSG00000088916 0
## ENSMUSG00000046101 0
## ENSMUSG00000098234 17# 2. El método específico para accesar la matriz de cuentas "counts"
counts(sce)[110:115, 1:3]## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 0
## ENSMUSG00000025915 8
## ENSMUSG00000046334 2
## ENSMUSG00000088916 0
## ENSMUSG00000046101 0
## ENSMUSG00000098234 63
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 0
## ENSMUSG00000025915 0
## ENSMUSG00000046334 3
## ENSMUSG00000088916 0
## ENSMUSG00000046101 0
## ENSMUSG00000098234 21
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 19
## ENSMUSG00000025915 1
## ENSMUSG00000046334 15
## ENSMUSG00000088916 0
## ENSMUSG00000046101 0
## ENSMUSG00000098234 17# AGREGAR MAS ASSAYS
sce <- scater::logNormCounts(sce)
# Revisa el objeto que acabamos de actualizar
sce## class: SingleCellExperiment
## dim: 46604 192
## metadata(0):
## assays(2): counts logcounts
## rownames(46604): ENSMUSG00000102693 ENSMUSG00000064842 ...
## ENSMUSG00000095742 CBFB-MYH11-mcherry
## rowData names(0):
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(1): sizeFactor
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce) / 1024^2 ## En MB## 106.5302 B# 1. El método general
assay(sce, "logcounts")[110:115, 1:3]## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 0.000000
## ENSMUSG00000025915 3.557154
## ENSMUSG00000046334 1.884687
## ENSMUSG00000088916 0.000000
## ENSMUSG00000046101 0.000000
## ENSMUSG00000098234 6.423258
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 0.000000
## ENSMUSG00000025915 0.000000
## ENSMUSG00000046334 2.087367
## ENSMUSG00000088916 0.000000
## ENSMUSG00000046101 0.000000
## ENSMUSG00000098234 4.569735
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 4.3052563
## ENSMUSG00000025915 0.9912494
## ENSMUSG00000046334 3.9835488
## ENSMUSG00000088916 0.0000000
## ENSMUSG00000046101 0.0000000
## ENSMUSG00000098234 4.1533513# 2. El método específico para accesar la matriz de cuentas
# transformadas "logcounts"
logcounts(sce)[110:115, 1:3]## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 0.000000
## ENSMUSG00000025915 3.557154
## ENSMUSG00000046334 1.884687
## ENSMUSG00000088916 0.000000
## ENSMUSG00000046101 0.000000
## ENSMUSG00000098234 6.423258
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 0.000000
## ENSMUSG00000025915 0.000000
## ENSMUSG00000046334 2.087367
## ENSMUSG00000088916 0.000000
## ENSMUSG00000046101 0.000000
## ENSMUSG00000098234 4.569735
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1
## ENSMUSG00000097893 4.3052563
## ENSMUSG00000025915 0.9912494
## ENSMUSG00000046334 3.9835488
## ENSMUSG00000088916 0.0000000
## ENSMUSG00000046101 0.0000000
## ENSMUSG00000098234 4.1533513# agregemos un assay mas, esta vez de manera manual
assay(sce, "counts_100") <- assay(sce, "counts") + 100 # suma 100 a counts assay
# Enumera los "assays" en el objeto
assays(sce) # indica num y nombre de assays## List of length 3
## names(3): counts logcounts counts_100assayNames(sce) # solo nos dará los nombres de los assays## [1] "counts" "logcounts" "counts_100"# assay(sce, "counts_100")[110:115, 1:3]
## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce) / 1024^2 ## En MB## 174.7981 B"segunda parte:
aquí checaremos metadata de las células"## [1] "segunda parte:\naquí checaremos metadata de las células"# Extrae la información de las muestras (metadata) del set de datos de 416b
colData.416b <- colData(sce.416b) # podemos checar objeto en la cajita de environment de RStudio!!
# explorar datooos
table(colData.416b$phenotype)##
## induced CBFB-MYH11 oncogene expression wild type phenotype
## 96 96table(colData.416b$block) # fue en varios dias?##
## 20160113 20160325
## 96 96# Agrega algo de esa información a nuestro objeto de SCE
colData(sce) <- colData.416b[, c("phenotype", "block")]
# Revisa el objeto que acabamos de actualizar
sce## class: SingleCellExperiment
## dim: 46604 192
## metadata(0):
## assays(3): counts logcounts counts_100
## rownames(46604): ENSMUSG00000102693 ENSMUSG00000064842 ...
## ENSMUSG00000095742 CBFB-MYH11-mcherry
## rowData names(0):
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(2): phenotype block
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):# Accesa a la información de las muestras (metadata) en nuestro SCE
colData(sce) # usar head?## DataFrame with 192 rows and 2 columns
## phenotype block
## <character> <integer>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 wild type phenotype 20160113
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 wild type phenotype 20160113
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 wild type phenotype 20160113
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. 20160113
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. 20160113
## ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 induced CBFB-MYH11 o.. 20160325
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 induced CBFB-MYH11 o.. 20160325
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 induced CBFB-MYH11 o.. 20160325
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 induced CBFB-MYH11 o.. 20160325
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 wild type phenotype 20160325# Accesa una columna específica de la información de las muestras (metadata)
table(sce$block)##
## 20160113 20160325
## 96 96table(colData(sce)$block) # otra manera##
## 20160113 20160325
## 96 96# Ejemplo de una función que agrega columnas nuevas al colData
sce <- scater::addPerCellQC(sce.416b) # añade datos de control de calidad
# Accesa a la información de las muestras (metadata) en nuestro SCE actualizado
colData(sce)## DataFrame with 192 rows and 18 columns
## Source Name cell line
## <character> <character>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 SLX-9555.N701_S502.C.. 416B
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 SLX-9555.N701_S503.C.. 416B
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 SLX-9555.N701_S504.C.. 416B
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 SLX-9555.N701_S505.C.. 416B
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 SLX-9555.N701_S506.C.. 416B
## ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S505... 416B
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S506... 416B
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S507... 416B
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S508... 416B
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 SLX-11312.N712_S517... 416B
## cell type
## <character>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 embryonic stem cell
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 embryonic stem cell
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 embryonic stem cell
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 embryonic stem cell
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 embryonic stem cell
## ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 embryonic stem cell
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 embryonic stem cell
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 embryonic stem cell
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 embryonic stem cell
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 embryonic stem cell
## single cell well quality
## <character>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 OK
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 OK
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 OK
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 OK
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 OK
## ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 OK
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 OK
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 OK
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 OK
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 OK
## genotype
## <character>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 Doxycycline-inducibl..
## ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl..
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl..
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl..
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl..
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 Doxycycline-inducibl..
## phenotype strain
## <character> <character>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 wild type phenotype B6D2F1-J
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 wild type phenotype B6D2F1-J
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 wild type phenotype B6D2F1-J
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. B6D2F1-J
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 induced CBFB-MYH11 o.. B6D2F1-J
## ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 induced CBFB-MYH11 o.. B6D2F1-J
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 induced CBFB-MYH11 o.. B6D2F1-J
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 induced CBFB-MYH11 o.. B6D2F1-J
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 induced CBFB-MYH11 o.. B6D2F1-J
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 wild type phenotype B6D2F1-J
## spike-in addition block sum
## <character> <integer> <numeric>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 ERCC+SIRV 20160113 865936
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ERCC+SIRV 20160113 1076277
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 ERCC+SIRV 20160113 1180138
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 ERCC+SIRV 20160113 1342593
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 ERCC+SIRV 20160113 1668311
## ... ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 Premixed 20160325 776622
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 Premixed 20160325 1299950
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 Premixed 20160325 1800696
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 Premixed 20160325 46731
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 Premixed 20160325 1866692
## detected altexps_ERCC_sum
## <numeric> <numeric>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 7618 65278
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 7521 74748
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 8306 60878
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 8143 60073
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 7154 136810
## ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 8174 61575
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 8956 94982
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 9530 113707
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 6649 7580
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 10964 48664
## altexps_ERCC_detected
## <numeric>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 39
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 40
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 42
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 42
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 44
## ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 39
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 41
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 40
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 44
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 39
## altexps_ERCC_percent altexps_SIRV_sum
## <numeric> <numeric>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 6.80658 27828
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 6.28030 39173
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 4.78949 30058
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 4.18567 32542
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 7.28887 71850
## ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 7.17620 19848
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 6.65764 31729
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 5.81467 41116
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 13.48898 1883
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 2.51930 16289
## altexps_SIRV_detected
## <numeric>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 7
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 7
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 7
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 7
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 7
## ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 7
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 7
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 7
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 7
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 7
## altexps_SIRV_percent total
## <numeric> <numeric>
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 2.90165 959042
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 3.29130 1190198
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 2.36477 1271074
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 2.26741 1435208
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 3.82798 1876971
## ... ... ...
## SLX-11312.N712_S505.H5H5YBBXX.s_8.r_1 2.313165 858045
## SLX-11312.N712_S506.H5H5YBBXX.s_8.r_1 2.224004 1426661
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1 2.102562 1955519
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1 3.350892 56194
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1 0.843271 1931645# Revisa el objeto que acabamos de actualizar
sce## class: SingleCellExperiment
## dim: 46604 192
## metadata(0):
## assays(1): counts
## rownames(46604): ENSMUSG00000102693 ENSMUSG00000064842 ...
## ENSMUSG00000095742 CBFB-MYH11-mcherry
## rowData names(1): Length
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(18): Source Name cell line ... altexps_SIRV_percent total
## reducedDimNames(0):
## mainExpName: endogenous
## altExpNames(2): ERCC SIRV## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce) / 1024^2 ## En MB## 39.47633 B## Agrega las cuentas normalizadas (lognorm) de nuevo
sce <- scater::logNormCounts(sce)
## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce) / 1024^2 ## En MB## 107.7463 B# Ejemplo: obtén el subconjunto de células de fenotipo "wild type"
# Acuérdate que las células son columnas del SCE !!!!
sce[, sce$phenotype == "wild type phenotype"]## class: SingleCellExperiment
## dim: 46604 96
## metadata(0):
## assays(2): counts logcounts
## rownames(46604): ENSMUSG00000102693 ENSMUSG00000064842 ...
## ENSMUSG00000095742 CBFB-MYH11-mcherry
## rowData names(1): Length
## colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S504.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(19): Source Name cell line ... total sizeFactor
## reducedDimNames(0):
## mainExpName: endogenous
## altExpNames(2): ERCC SIRV"Tercera parte:
examinaremos metadata de features (rowData)"## [1] "Tercera parte:\nexaminaremos metadata de features (rowData)"# Accesa la información de los genes de nuestro SCE
# ¡Está vació actualmente!
rowData(sce)## DataFrame with 46604 rows and 1 column
## Length
## <integer>
## ENSMUSG00000102693 1070
## ENSMUSG00000064842 110
## ENSMUSG00000051951 6094
## ENSMUSG00000102851 480
## ENSMUSG00000103377 2819
## ... ...
## ENSMUSG00000094621 121
## ENSMUSG00000098647 99
## ENSMUSG00000096730 3077
## ENSMUSG00000095742 243
## CBFB-MYH11-mcherry 2998# Ejemplo de una función que agrega campos nuevos en el rowData
sce <- scater::addPerFeatureQC(sce)
# Accesa a la información de las muestras (metadata) en nuestro SCE actualizado
rowData(sce)## DataFrame with 46604 rows and 3 columns
## Length mean detected
## <integer> <numeric> <numeric>
## ENSMUSG00000102693 1070 0.0000000 0.000000
## ENSMUSG00000064842 110 0.0000000 0.000000
## ENSMUSG00000051951 6094 0.0000000 0.000000
## ENSMUSG00000102851 480 0.0000000 0.000000
## ENSMUSG00000103377 2819 0.0104167 0.520833
## ... ... ... ...
## ENSMUSG00000094621 121 0.0 0
## ENSMUSG00000098647 99 0.0 0
## ENSMUSG00000096730 3077 0.0 0
## ENSMUSG00000095742 243 0.0 0
## CBFB-MYH11-mcherry 2998 50375.7 100## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce) / 1024^2 ## En MB## 108.4578 B# Descarga los archivos de anotación de la base de datos de Ensembl
# correspondientes usando los recursos disponibles vía AnnotationHub
library("AnnotationHub")
ah <- AnnotationHub()
query(ah, c("Mus musculus", "Ensembl", "v97"))## AnnotationHub with 1 record
## # snapshotDate(): 2021-05-18
## # names(): AH73905
## # $dataprovider: Ensembl
## # $species: Mus musculus
## # $rdataclass: EnsDb
## # $rdatadateadded: 2019-05-02
## # $title: Ensembl 97 EnsDb for Mus musculus
## # $description: Gene and protein annotations for Mus musculus based on Ensem...
## # $taxonomyid: 10090
## # $genome: GRCm38
## # $sourcetype: ensembl
## # $sourceurl: http://www.ensembl.org
## # $sourcesize: NA
## # $tags: c("97", "AHEnsDbs", "Annotation", "EnsDb", "Ensembl", "Gene",
## # "Protein", "Transcript")
## # retrieve record with 'object[["AH73905"]]'# Obtén la posición del cromosoma para cada gen
ensdb <- ah[["AH73905"]]
chromosome <- mapIds(ensdb,
keys = rownames(sce),
keytype = "GENEID",
column = "SEQNAME"
)
rowData(sce)$chromosome <- chromosome
# Accesa a la información de las muestras (metadata) en nuestro SCE actualizado
rowData(sce)## DataFrame with 46604 rows and 4 columns
## Length mean detected chromosome
## <integer> <numeric> <numeric> <character>
## ENSMUSG00000102693 1070 0.0000000 0.000000 1
## ENSMUSG00000064842 110 0.0000000 0.000000 1
## ENSMUSG00000051951 6094 0.0000000 0.000000 1
## ENSMUSG00000102851 480 0.0000000 0.000000 1
## ENSMUSG00000103377 2819 0.0104167 0.520833 1
## ... ... ... ... ...
## ENSMUSG00000094621 121 0.0 0 GL456372.1
## ENSMUSG00000098647 99 0.0 0 GL456381.1
## ENSMUSG00000096730 3077 0.0 0 JH584292.1
## ENSMUSG00000095742 243 0.0 0 JH584295.1
## CBFB-MYH11-mcherry 2998 50375.7 100 NA## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce) / 1024^2 ## En MB## 108.8135 B# Ejemplo: obtén el subconjunto de datos donde los genes están en el
# cromosoma 3
# NOTA: which() fue necesario para lidear con los nombres de cromosoma
# que son NA
sce[which(rowData(sce)$chromosome == "3"), ]## class: SingleCellExperiment
## dim: 2876 192
## metadata(0):
## assays(2): counts logcounts
## rownames(2876): ENSMUSG00000098982 ENSMUSG00000098307 ...
## ENSMUSG00000105990 ENSMUSG00000075903
## rowData names(4): Length mean detected chromosome
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(19): Source Name cell line ... total sizeFactor
## reducedDimNames(0):
## mainExpName: endogenous
## altExpNames(2): ERCC SIRV"Cuarta parte:
examinamos slot metadata"## [1] "Cuarta parte:\nexaminamos slot metadata"# Accesa la información de nuestro experimento usando metadata()
# ¡Está vació actualmente!
metadata(sce)## list()# La información en el metadata() es como Vegas - todo se vale
metadata(sce) <- list(
favourite_genes = c("Shh", "Nck1", "Diablo"),
analyst = c("Pete")
)
# Accesa la información de nuestro experimento usando metadata() de
# nuestro objeto actualizado
metadata(sce)## $favourite_genes
## [1] "Shh" "Nck1" "Diablo"
##
## $analyst
## [1] "Pete""Quinta parte:
examinamos slot de reducción de dimensiones"## [1] "Quinta parte:\nexaminamos slot de reducción de dimensiones"# Ejemplo: agrega los componentes principales (PCs) de las logcounts
# NOTA: aprenderemos más sobre análisis de componentes principales (PCA) después
sce <- scater::runPCA(sce)
# Revisa el objeto que acabamos de actualizar
sce## class: SingleCellExperiment
## dim: 46604 192
## metadata(2): favourite_genes analyst
## assays(2): counts logcounts
## rownames(46604): ENSMUSG00000102693 ENSMUSG00000064842 ...
## ENSMUSG00000095742 CBFB-MYH11-mcherry
## rowData names(4): Length mean detected chromosome
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(19): Source Name cell line ... total sizeFactor
## reducedDimNames(1): PCA
## mainExpName: endogenous
## altExpNames(2): ERCC SIRV# Accesa la matriz de PCA del componente (slot) reducedDims
reducedDim(sce, "PCA")[1:6, 1:3]## PC1 PC2 PC3
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 -18.717668 27.598132 -5.939654
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 -2.480705 27.564583 -4.916567
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 -42.034018 7.552435 -12.126964
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 8.494303 -31.833727 -15.760853
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 49.737390 -4.226795 -6.123169
## SLX-9555.N701_S507.C89V9ANXX.s_1.r_1 44.528081 3.215503 -10.384939# Ejemplo, agrega una representación de los logcounts en t-SNE
# NOTA: aprenderemos más sobre t-SNE después
sce <- scater::runTSNE(sce)
# Revisa el objeto que acabamos de actualizar
sce## class: SingleCellExperiment
## dim: 46604 192
## metadata(2): favourite_genes analyst
## assays(2): counts logcounts
## rownames(46604): ENSMUSG00000102693 ENSMUSG00000064842 ...
## ENSMUSG00000095742 CBFB-MYH11-mcherry
## rowData names(4): Length mean detected chromosome
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(19): Source Name cell line ... total sizeFactor
## reducedDimNames(2): PCA TSNE
## mainExpName: endogenous
## altExpNames(2): ERCC SIRV# Accesa a la matriz de t-SNE en el componente (slot) de reducedDims
head(reducedDim(sce, "TSNE"))## [,1] [,2]
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 -7.3181642 2.2840490
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 -3.6464118 -0.8207964
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 -7.6479076 4.6719923
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 0.7775377 4.4127326
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 8.0179911 -6.4277429
## SLX-9555.N701_S507.C89V9ANXX.s_1.r_1 7.8965473 -7.8963465# Ejemplo: agrega una representación 'manual' de los logcounts en UMAP
# NOTA: aprenderemos más sobre UMAP después y de una forma más sencilla de
# calcularla
u <- uwot::umap(t(logcounts(sce)), n_components = 2)
# Agrega la matriz de UMAP al componente (slot) reducedDims
reducedDim(sce, "UMAP") <- u
# Accesa a la matriz de UMAP desde el componente (slot) reducedDims
head(reducedDim(sce, "UMAP"))## [,1] [,2]
## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 -2.9007656 -1.724959
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 -1.9364034 -1.711854
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 -3.2449543 -1.109732
## SLX-9555.N701_S505.C89V9ANXX.s_1.r_1 -0.3859376 -1.198746
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 0.2112636 -1.497831
## SLX-9555.N701_S507.C89V9ANXX.s_1.r_1 -0.1741835 -1.455346# Enumera los resultados de reducción de dimensiones en nuestro objeto SCE
reducedDims(sce)## List of length 3
## names(3): PCA TSNE UMAP"Sexta parte:
experimentos alternativos"## [1] "Sexta parte:\nexperimentos alternativos"# Extrae la información de ERCC de nuestro SCE para el set de datos de 416b
ercc.sce.416b <- altExp(sce.416b, "ERCC")
# Inspecciona el SCE para los datos de ERCC
ercc.sce.416b## class: SingleCellExperiment
## dim: 92 192
## metadata(0):
## assays(1): counts
## rownames(92): ERCC-00002 ERCC-00003 ... ERCC-00170 ERCC-00171
## rowData names(7): Length subgroup ... log2(Mix 1/Mix 2) molecules
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(0):
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):# Agrega el SCE de ERCC como un experimento alternativo a nuestro SCE
altExp(sce, "ERCC") <- ercc.sce.416b
# Revisa el objeto que acabamos de actualizar
sce## class: SingleCellExperiment
## dim: 46604 192
## metadata(2): favourite_genes analyst
## assays(2): counts logcounts
## rownames(46604): ENSMUSG00000102693 ENSMUSG00000064842 ...
## ENSMUSG00000095742 CBFB-MYH11-mcherry
## rowData names(4): Length mean detected chromosome
## colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(19): Source Name cell line ... total sizeFactor
## reducedDimNames(3): PCA TSNE UMAP
## mainExpName: endogenous
## altExpNames(2): ERCC SIRV## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce) / 1024^2 ## En MB## 108.8965 B# Enumera los experimentos alternativos almacenados en nuestro objeto
altExps(sce)## List of length 2
## names(2): ERCC SIRV# El crear un subconjunto del SCE por muestra (célula) automáticamente
# obtiene el subconjunto de los experimentos alternativos
sce.subset <- sce[, 1:10]
ncol(sce.subset)## [1] 10ncol(altExp(sce.subset))## [1] 10## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce.subset) / 1024^2 ## En MB## 11.99544 B"Septima parte:
sizefactores"## [1] "Septima parte:\nsizefactores"# Extrae los factores de tamaño (size factors)
# Estos fueron añadidos a nuestro objeto cuando corrimos
# scater::logNormCounts(sce)
head(sizeFactors(sce))## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
## 0.7427411 0.9231573
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 SLX-9555.N701_S505.C89V9ANXX.s_1.r_1
## 1.0122422 1.1515851
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 SLX-9555.N701_S507.C89V9ANXX.s_1.r_1
## 1.4309639 0.8713409# "Automáticamente" reemplaza los factores de tamaño
sce <- scran::computeSumFactors(sce)
head(sizeFactors(sce))## [1] 0.6961756 0.8834223 0.9704247 0.9804890 1.2446699 0.7922620# "Manualmente" reemplaza los factores de tamaño
sizeFactors(sce) <- scater::librarySizeFactors(sce)
head(sizeFactors(sce))## SLX-9555.N701_S502.C89V9ANXX.s_1.r_1 SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
## 0.7427411 0.9231573
## SLX-9555.N701_S504.C89V9ANXX.s_1.r_1 SLX-9555.N701_S505.C89V9ANXX.s_1.r_1
## 1.0122422 1.1515851
## SLX-9555.N701_S506.C89V9ANXX.s_1.r_1 SLX-9555.N701_S507.C89V9ANXX.s_1.r_1
## 1.4309639 0.8713409NOTA: La clase SingleCellExperimentestá basada en SummarizedExperiment, por lo que ya estamos un poco familiarizados con esta nueva clase.
Una de las diferencias es que no contiene ranuras para análisis de reducción de dimensiones. (será solo datos y metadata de ellos)
4.3.1 Preguntas:
¿Cuáles son los tipos de tablas que debe siempre contenter el objeto
sce?¿Donde usamos los
colnames(sce)?¿donde usamos los
rownames(sce)?
4.4 Breve repaso de R
# Creamos un data.frame
df <- data.frame(x = c(TRUE, FALSE, NA, NA), y = c(12, 34, 56, 78))
row.names(df) <- letters[1:4]
df## x y
## a TRUE 12
## b FALSE 34
## c NA 56
## d NA 78# Para acceder a los nombres de las columnas
colnames(df)## [1] "x" "y"# Para acceder a los nombres de las filas
rownames(df)## [1] "a" "b" "c" "d"# Podemos sacar información booleana
df$y < 20## [1] TRUE FALSE FALSE FALSE# Y podemos acceder al mismo data.frame con la información booleana
df[df$y < 40, ]## x y
## a TRUE 12
## b FALSE 34## %in% (dentro de)
bool_info <- rownames(df) %in% c("a", "c", "z")
df[bool_info, ]## x y
## a TRUE 12
## c NA 56## & (y)
bool_info <- df$y < 50 & df$y > 20
df[bool_info, ]## x y
## b FALSE 34## | (o)
bool_info <- df$y < 20 | df$y > 60
df[bool_info, ]## x y
## a TRUE 12
## d NA 784.5 Ejercicio 2
library("SingleCellExperiment")
library("scRNAseq")
# Mini muestreo del set de datos usado en: https://bioconductor.org/books/release/OSCA/zeisel-mouse-brain-strt-seq.html#introduction-5
archivo_cuentas <- "https://raw.githubusercontent.com/emarquezz/minidataset_osca/main/min_sce.csv"
archivo_rowData <- "https://raw.githubusercontent.com/emarquezz/minidataset_osca/main/rowD.csv"
archivo_colData <- "https://raw.githubusercontent.com/emarquezz/minidataset_osca/main/colD.csv"
counts <- read.csv(archivo_cuentas, row.names = 1, header = TRUE, check.names = F)
col.data <- DataFrame(read.csv(archivo_colData, row.names = 1, header = TRUE, check.names = F))
row.data <- read.csv(archivo_rowData, row.names = 1, header = TRUE, check.names = F)Crea un objeto
SingleCellExperiment¿Cuántos genes tenemos?
¿Qué información tenemos en el
rowData()?
Extraigan los datos de los genes que nos interesan (objeto
int_gen)Pasen el mouse sobre los siguientes textos para ver recomendaciones:
int_gen <- c("Angpt1", "Chic2", "Mir503", "Magee2", "Nenf", "Eps15l1", "Hsf2bp", "Gnptg", "Vegfb", "Atmin", "Gad1", "Gad2", "Slc32a1", "Dner", "Slc2a13", "Slc6a1", "Nrxn3")Creen un objeto llamado
min_scecon los datos de solo esos genes¿Cuáles son parte del tejido
interneuronso del tejidopyramidal CA1? (del objetomin_sce)
Con este subconjunto, crea el objeto
tej_min_sceUna vez que tengan el objeto ´SingleCellExperiment´ llamado ´tej_min_sce´, corran el siguiente código.
library("scater")
plotHeatmap(object = tej_min_sce, features = rownames(tej_min_sce), order_columns_by = "level1class")4.5.1 Respuestas
sce <- SingleCellExperiment(
assays = list(counts = counts),
colData = col.data,
rowData = row.data
)
sce <- scater::logNormCounts(sce)
sce## class: SingleCellExperiment
## dim: 100 30
## metadata(0):
## assays(2): counts logcounts
## rownames(100): Olfr1219 Angpt1 ... Slc6a1 Nrxn3
## rowData names(2): featureType Ensembl
## colnames(30): 1772066102_E02 1772067073_F10 ... 1772066096_G11
## 1772066101_E11
## colData names(11): tissue group.. ... level2class sizeFactor
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):bool_data <- rownames(rowData(sce))
min_sce <- sce[bool_data %in% int_gen, ]tej_int <- min_sce$level1class == "interneurons"
tej_pyr <- min_sce$level1class == "pyramidal CA1"
tej_min_sce <- min_sce[, tej_int | tej_pyr]library("scater")## Loading required package: scuttle## Loading required package: ggplot2plotHeatmap(object = tej_min_sce, features = rownames(tej_min_sce), order_columns_by = "level1class")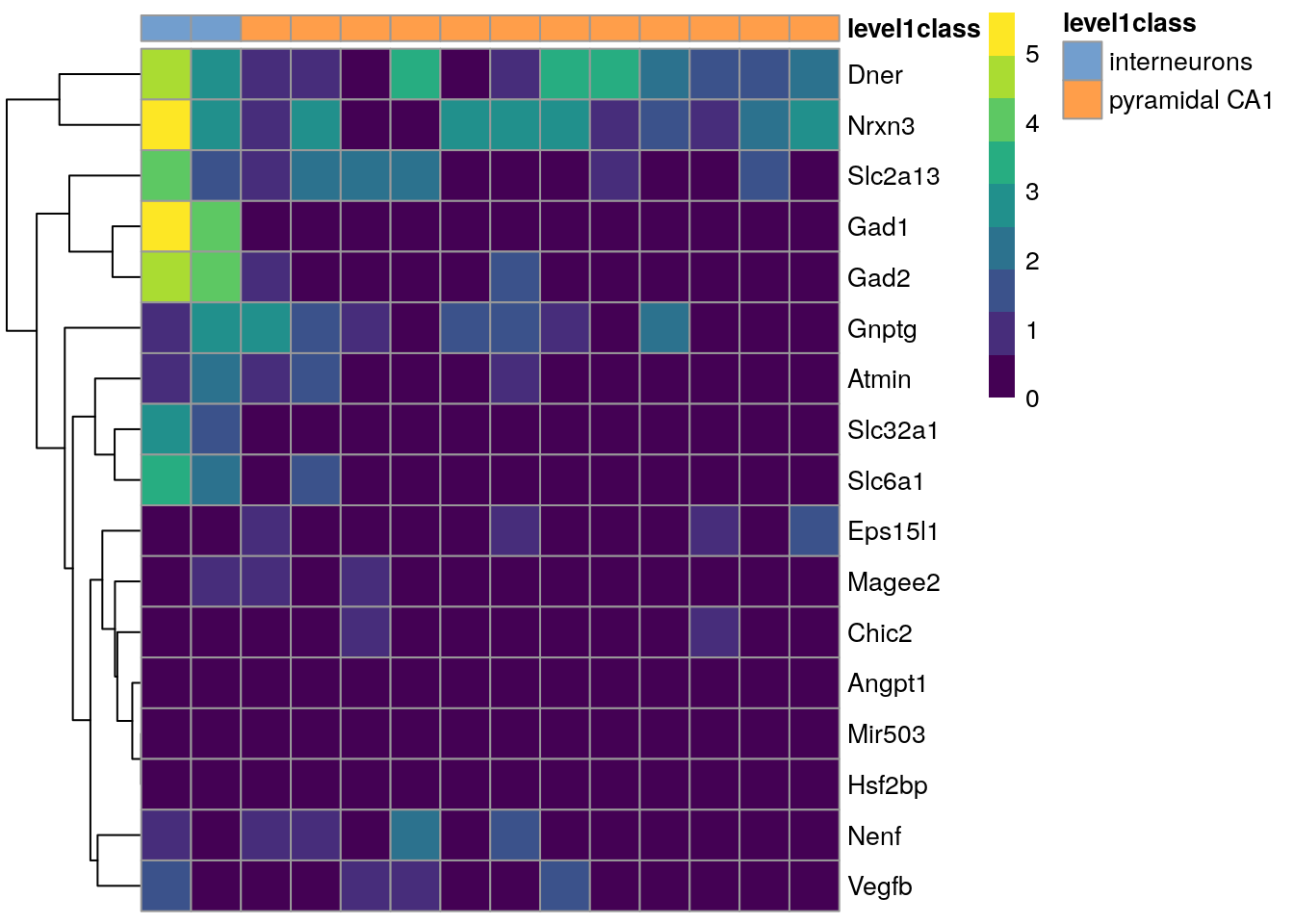
4.5.2 Extra
- Realiza los mismos pasos, pero ahora los genes que buscamos no tienen el nombre usual de gen (Gad1), sino su Ensembl gene IDs
int_gen <- c("ENSMUSG00000071076", "ENSMUSG00000002076", "ENSMUSG00000024962", "ENSMUSG00000031224", "ENSMSG00000036560", "ENSMUSG00000037499", "ENSMUSG00000006276", "ENSMUSG00000035521", "ENSMUSG00000047388", "ENSMUSG0000051079", "ENSMUSG00000076122", "ENSMUSG00000029229", "ENSMUSG00000022309", "ENSMUSG00000036766", "ENSMUSG00000070880", "ENSMUSG00000026787", "ENSMUSG00000066392", "ENSMUSG00000036298", "ENSMUSG00000037771", "ENSMUSG00000030310")4.6 Import data
- Diapositivas de Peter Hickey
Ve las diapositivas aquí.
# Descarga datos de ejemplo procesados con CellRanger
# Paréntesis: al usar BiocFileCache solo tenemos que descargar
# los datos una vez.
library("BiocFileCache")
bfc <- BiocFileCache()
pbmc.url <-
paste0(
"http://cf.10xgenomics.com/samples/cell-vdj/",
"3.1.0/vdj_v1_hs_pbmc3/",
"vdj_v1_hs_pbmc3_filtered_feature_bc_matrix.tar.gz"
)
pbmc.data <- bfcrpath(bfc, pbmc.url)## adding rname 'http://cf.10xgenomics.com/samples/cell-vdj/3.1.0/vdj_v1_hs_pbmc3/vdj_v1_hs_pbmc3_filtered_feature_bc_matrix.tar.gz'# Extrae los archivos en un directorio temporal
untar(pbmc.data, exdir = tempdir())
# Enumera los archivos que descargamos y que extrajimos
# Estos son los archivos típicos de CellRanger
pbmc.dir <- file.path(
tempdir(),
"filtered_feature_bc_matrix"
)
list.files(pbmc.dir)## [1] "barcodes.tsv.gz" "features.tsv.gz" "matrix.mtx.gz"# Importa los datos como un objeto de tipo SingleCellExperiment
library("DropletUtils")
sce.pbmc <- read10xCounts(pbmc.dir)
# Revisa el objeto que acabamos de construir
sce.pbmc## class: SingleCellExperiment
## dim: 33555 7231
## metadata(1): Samples
## assays(1): counts
## rownames(33555): ENSG00000243485 ENSG00000237613 ... CD127 CD15
## rowData names(3): ID Symbol Type
## colnames: NULL
## colData names(2): Sample Barcode
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce.pbmc) / 1024^2 ## En MB## 124.191 B# Almacena la información de CITE-seq como un experimento alternativo
sce.pbmc <- splitAltExps(sce.pbmc, rowData(sce.pbmc)$Type)
# Revisa el objeto que acabamos de actualizar
sce.pbmc## class: SingleCellExperiment
## dim: 33538 7231
## metadata(1): Samples
## assays(1): counts
## rownames(33538): ENSG00000243485 ENSG00000237613 ... ENSG00000277475
## ENSG00000268674
## rowData names(3): ID Symbol Type
## colnames: NULL
## colData names(2): Sample Barcode
## reducedDimNames(0):
## mainExpName: Gene Expression
## altExpNames(1): Antibody Capture## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce.pbmc) / 1024^2 ## En MB## 124.4871 B# Descarga datos de ejemplo procesados con scPipe
library("BiocFileCache")
bfc <- BiocFileCache()
sis_seq.url <-
"https://github.com/LuyiTian/SIS-seq_script/archive/master.zip"
sis_seq.data <- bfcrpath(bfc, sis_seq.url)## adding rname 'https://github.com/LuyiTian/SIS-seq_script/archive/master.zip'# Extrae los archivos en un directorio temporal
unzip(sis_seq.data, exdir = tempdir())
# Enumera (algunos de) los archivos que descargamos y extrajimos
# Estos son los archivos típicos de scPipe
sis_seq.dir <- file.path(
tempdir(),
"SIS-seq_script-master",
"data",
"BcorKO_scRNAseq",
"RPI10"
)
list.files(sis_seq.dir)## [1] "gene_count.csv" "stat"# Importa los datos como un objeto de tipo SingleCellExperiment
library("scPipe")## ## ## Registered S3 method overwritten by 'GGally':
## method from
## +.gg ggplot2sce.sis_seq <- create_sce_by_dir(sis_seq.dir)## organism/gene_id_type not provided. Make a guess: mmusculus_gene_ensembl / ensembl_gene_id# Revisa el objeto que acabamos de construir
sce.sis_seq## class: SingleCellExperiment
## dim: 19232 383
## metadata(2): scPipe Biomart
## assays(1): counts
## rownames(19232): ENSMUSG00000079140 ENSMUSG00000081587 ...
## ENSMUSG00000036880 ENSMUSG00000106872
## rowData names(0):
## colnames(383): A1 A10 ... P8 P9
## colData names(7): unaligned aligned_unmapped ... mapped_to_ERCC
## mapped_to_MT
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(sce.sis_seq) / 1024^2 ## En MB## 29.84988 B# Descarga un ejemplo de un montón de archivos
library("BiocFileCache")
bfc <- BiocFileCache()
lun_counts.url <-
paste0(
"https://www.ebi.ac.uk/arrayexpress/files/",
"E-MTAB-5522/E-MTAB-5522.processed.1.zip"
)
lun_counts.data <- bfcrpath(bfc, lun_counts.url)## adding rname 'https://www.ebi.ac.uk/arrayexpress/files/E-MTAB-5522/E-MTAB-5522.processed.1.zip'lun_coldata.url <-
paste0(
"https://www.ebi.ac.uk/arrayexpress/files/",
"E-MTAB-5522/E-MTAB-5522.sdrf.txt"
)
lun_coldata.data <- bfcrpath(bfc, lun_coldata.url)## adding rname 'https://www.ebi.ac.uk/arrayexpress/files/E-MTAB-5522/E-MTAB-5522.sdrf.txt'# Extrae los archivos en un directorio temporal
lun_counts.dir <- tempfile("lun_counts.")
unzip(lun_counts.data, exdir = lun_counts.dir)
# Enumera los archivos que descargamos y extrajimos
list.files(lun_counts.dir)## [1] "counts_Calero_20160113.tsv" "counts_Calero_20160325.tsv"
## [3] "counts_Liora_20160906.tsv" "counts_Liora_20170201.tsv"# Lee la matriz de cuentas (para una placa)
lun.counts <- read.delim(
file.path(lun_counts.dir, "counts_Calero_20160113.tsv"),
header = TRUE,
row.names = 1,
check.names = FALSE
)
# Almacena la información de la longitud de los genes para después
gene.lengths <- lun.counts$Length
# Convierte los datos de cuentas de genez a una matriz (quitamos las longitudes)
lun.counts <- as.matrix(lun.counts[, -1])
# Lee la información de las muestras (células)
lun.coldata <- read.delim(lun_coldata.data,
check.names = FALSE,
stringsAsFactors = FALSE
)
library("S4Vectors")
lun.coldata <- as(lun.coldata, "DataFrame")
# Pon en orden la información de las muestras para que
# sea idéntico al orden en la matriz de cuentas
m <- match(
colnames(lun.counts),
lun.coldata$`Source Name`
)
lun.coldata <- lun.coldata[m, ]
# Construye la tabla de información de los genes
lun.rowdata <- DataFrame(Length = gene.lengths)
# Construye el objeto de SingleCellExperiment
lun.sce <- SingleCellExperiment(
assays = list(assays = lun.counts),
colData = lun.coldata,
rowData = lun.rowdata
)
# Revisa el objeto que acabamos de construir
lun.sce## class: SingleCellExperiment
## dim: 46703 96
## metadata(0):
## assays(1): assays
## rownames(46703): ENSMUSG00000102693 ENSMUSG00000064842 ... SIRV7
## CBFB-MYH11-mcherry
## rowData names(1): Length
## colnames(96): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-9555.N712_S508.C89V9ANXX.s_1.r_1
## SLX-9555.N712_S517.C89V9ANXX.s_1.r_1
## colData names(50): Source Name Comment[ENA_SAMPLE] ... Factor
## Value[phenotype] Factor Value[block]
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):## ¿Qué tan grande es el objeto de R?
lobstr::obj_size(lun.sce) / 1024^2 ## En MB## 21.49744 B4.7 Detalles de la sesión de R
## Información de la sesión de R
Sys.time()## [1] "2021-08-19 15:17:33 UTC"proc.time()## user system elapsed
## 75.942 4.503 105.138options(width = 120)
sessioninfo::session_info()## ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
## setting value
## version R version 4.1.0 (2021-05-18)
## os Ubuntu 20.04.2 LTS
## system x86_64, linux-gnu
## ui X11
## language (EN)
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz UTC
## date 2021-08-19
##
## ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
## package * version date lib source
## AnnotationDbi * 1.54.1 2021-06-08 [1] Bioconductor
## AnnotationFilter * 1.16.0 2021-05-19 [1] Bioconductor
## AnnotationHub * 3.0.1 2021-06-20 [1] Bioconductor
## assertthat 0.2.1 2019-03-21 [1] RSPM (R 4.1.0)
## beachmat 2.8.1 2021-08-12 [1] Bioconductor
## beeswarm 0.4.0 2021-06-01 [1] RSPM (R 4.1.0)
## Biobase * 2.52.0 2021-05-19 [1] Bioconductor
## BiocFileCache * 2.0.0 2021-05-19 [1] Bioconductor
## BiocGenerics * 0.38.0 2021-05-19 [1] Bioconductor
## BiocIO 1.2.0 2021-05-19 [1] Bioconductor
## BiocManager 1.30.16 2021-06-15 [1] RSPM (R 4.1.0)
## BiocNeighbors 1.10.0 2021-05-19 [1] Bioconductor
## BiocParallel 1.26.1 2021-07-04 [1] Bioconductor
## BiocSingular 1.8.1 2021-06-08 [1] Bioconductor
## BiocVersion 3.13.1 2021-03-19 [2] Bioconductor
## biomaRt 2.48.3 2021-08-15 [1] Bioconductor
## Biostrings 2.60.2 2021-08-05 [1] Bioconductor
## bit 4.0.4 2020-08-04 [1] RSPM (R 4.1.0)
## bit64 4.0.5 2020-08-30 [1] RSPM (R 4.1.0)
## bitops 1.0-7 2021-04-24 [1] RSPM (R 4.1.0)
## blob 1.2.2 2021-07-23 [1] RSPM (R 4.1.0)
## bluster 1.2.1 2021-05-27 [1] Bioconductor
## bookdown 0.23 2021-08-13 [1] RSPM (R 4.1.0)
## bslib 0.2.5.1 2021-05-18 [1] RSPM (R 4.1.0)
## cachem 1.0.5 2021-05-15 [2] RSPM (R 4.1.0)
## cli 3.0.1 2021-07-17 [2] RSPM (R 4.1.0)
## cluster 2.1.2 2021-04-17 [3] CRAN (R 4.1.0)
## colorspace 2.0-2 2021-06-24 [1] RSPM (R 4.1.0)
## crayon 1.4.1 2021-02-08 [2] RSPM (R 4.1.0)
## curl 4.3.2 2021-06-23 [2] RSPM (R 4.1.0)
## DBI 1.1.1 2021-01-15 [1] RSPM (R 4.1.0)
## dbplyr * 2.1.1 2021-04-06 [1] RSPM (R 4.1.0)
## DelayedArray 0.18.0 2021-05-19 [1] Bioconductor
## DelayedMatrixStats 1.14.2 2021-08-08 [1] Bioconductor
## DEoptimR 1.0-9 2021-05-24 [1] RSPM (R 4.1.0)
## digest 0.6.27 2020-10-24 [2] RSPM (R 4.1.0)
## dplyr 1.0.7 2021-06-18 [1] RSPM (R 4.1.0)
## dqrng 0.3.0 2021-05-01 [1] RSPM (R 4.1.0)
## DropletUtils * 1.12.2 2021-07-22 [1] Bioconductor
## edgeR 3.34.0 2021-05-19 [1] Bioconductor
## ellipsis 0.3.2 2021-04-29 [2] RSPM (R 4.1.0)
## ensembldb * 2.16.4 2021-08-05 [1] Bioconductor
## evaluate 0.14 2019-05-28 [2] RSPM (R 4.1.0)
## ExperimentHub 2.0.0 2021-05-19 [1] Bioconductor
## fansi 0.5.0 2021-05-25 [2] RSPM (R 4.1.0)
## farver 2.1.0 2021-02-28 [1] RSPM (R 4.1.0)
## fastmap 1.1.0 2021-01-25 [2] RSPM (R 4.1.0)
## filelock 1.0.2 2018-10-05 [1] RSPM (R 4.1.0)
## FNN 1.1.3 2019-02-15 [1] RSPM (R 4.1.0)
## generics 0.1.0 2020-10-31 [1] RSPM (R 4.1.0)
## GenomeInfoDb * 1.28.1 2021-07-01 [1] Bioconductor
## GenomeInfoDbData 1.2.6 2021-07-29 [1] Bioconductor
## GenomicAlignments 1.28.0 2021-05-19 [1] Bioconductor
## GenomicFeatures * 1.44.1 2021-08-15 [1] Bioconductor
## GenomicRanges * 1.44.0 2021-05-19 [1] Bioconductor
## GGally 2.1.2 2021-06-21 [1] RSPM (R 4.1.0)
## ggbeeswarm 0.6.0 2017-08-07 [1] RSPM (R 4.1.0)
## ggplot2 * 3.3.5 2021-06-25 [1] RSPM (R 4.1.0)
## glue 1.4.2 2020-08-27 [2] RSPM (R 4.1.0)
## gridExtra 2.3 2017-09-09 [1] RSPM (R 4.1.0)
## gtable 0.3.0 2019-03-25 [1] RSPM (R 4.1.0)
## HDF5Array 1.20.0 2021-05-19 [1] Bioconductor
## highr 0.9 2021-04-16 [2] RSPM (R 4.1.0)
## hms 1.1.0 2021-05-17 [1] RSPM (R 4.1.0)
## htmltools 0.5.1.1 2021-01-22 [1] RSPM (R 4.1.0)
## httpuv 1.6.1 2021-05-07 [1] RSPM (R 4.1.0)
## httr 1.4.2 2020-07-20 [2] RSPM (R 4.1.0)
## igraph 1.2.6 2020-10-06 [1] RSPM (R 4.1.0)
## interactiveDisplayBase 1.30.0 2021-05-19 [1] Bioconductor
## IRanges * 2.26.0 2021-05-19 [1] Bioconductor
## irlba 2.3.3 2019-02-05 [1] RSPM (R 4.1.0)
## jquerylib 0.1.4 2021-04-26 [1] RSPM (R 4.1.0)
## jsonlite 1.7.2 2020-12-09 [2] RSPM (R 4.1.0)
## KEGGREST 1.32.0 2021-05-19 [1] Bioconductor
## knitr 1.33 2021-04-24 [2] RSPM (R 4.1.0)
## later 1.2.0 2021-04-23 [1] RSPM (R 4.1.0)
## lattice 0.20-44 2021-05-02 [3] CRAN (R 4.1.0)
## lazyeval 0.2.2 2019-03-15 [1] RSPM (R 4.1.0)
## lifecycle 1.0.0 2021-02-15 [2] RSPM (R 4.1.0)
## limma 3.48.3 2021-08-10 [1] Bioconductor
## lobstr 1.1.1 2019-07-02 [1] RSPM (R 4.1.0)
## locfit 1.5-9.4 2020-03-25 [1] RSPM (R 4.1.0)
## magrittr 2.0.1 2020-11-17 [2] RSPM (R 4.1.0)
## Matrix 1.3-4 2021-06-01 [3] RSPM (R 4.1.0)
## MatrixGenerics * 1.4.2 2021-08-08 [1] Bioconductor
## matrixStats * 0.60.0 2021-07-26 [1] RSPM (R 4.1.0)
## mclust 5.4.7 2020-11-20 [1] RSPM (R 4.1.0)
## memoise 2.0.0 2021-01-26 [2] RSPM (R 4.1.0)
## metapod 1.0.0 2021-05-19 [1] Bioconductor
## mime 0.11 2021-06-23 [2] RSPM (R 4.1.0)
## munsell 0.5.0 2018-06-12 [1] RSPM (R 4.1.0)
## org.Hs.eg.db 3.13.0 2021-07-29 [1] Bioconductor
## org.Mm.eg.db 3.13.0 2021-07-29 [1] Bioconductor
## pheatmap 1.0.12 2019-01-04 [1] RSPM (R 4.1.0)
## pillar 1.6.2 2021-07-29 [2] RSPM (R 4.1.0)
## pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.1.0)
## plyr 1.8.6 2020-03-03 [1] RSPM (R 4.1.0)
## png 0.1-7 2013-12-03 [1] RSPM (R 4.1.0)
## prettyunits 1.1.1 2020-01-24 [2] RSPM (R 4.1.0)
## progress 1.2.2 2019-05-16 [1] RSPM (R 4.1.0)
## promises 1.2.0.1 2021-02-11 [1] RSPM (R 4.1.0)
## ProtGenerics 1.24.0 2021-05-19 [1] Bioconductor
## purrr 0.3.4 2020-04-17 [2] RSPM (R 4.1.0)
## R.methodsS3 1.8.1 2020-08-26 [1] RSPM (R 4.1.0)
## R.oo 1.24.0 2020-08-26 [1] RSPM (R 4.1.0)
## R.utils 2.10.1 2020-08-26 [1] RSPM (R 4.1.0)
## R6 2.5.0 2020-10-28 [2] RSPM (R 4.1.0)
## rappdirs 0.3.3 2021-01-31 [2] RSPM (R 4.1.0)
## RColorBrewer 1.1-2 2014-12-07 [1] RSPM (R 4.1.0)
## Rcpp 1.0.7 2021-07-07 [2] RSPM (R 4.1.0)
## RCurl 1.98-1.4 2021-08-17 [1] RSPM (R 4.1.0)
## reshape 0.8.8 2018-10-23 [1] RSPM (R 4.1.0)
## restfulr 0.0.13 2017-08-06 [1] RSPM (R 4.1.0)
## rhdf5 2.36.0 2021-05-19 [1] Bioconductor
## rhdf5filters 1.4.0 2021-05-19 [1] Bioconductor
## Rhdf5lib 1.14.2 2021-07-06 [1] Bioconductor
## Rhtslib 1.24.0 2021-05-19 [1] Bioconductor
## rjson 0.2.20 2018-06-08 [1] RSPM (R 4.1.0)
## rlang 0.4.11 2021-04-30 [2] RSPM (R 4.1.0)
## rmarkdown 2.10 2021-08-06 [1] RSPM (R 4.1.0)
## robustbase 0.93-8 2021-06-02 [1] RSPM (R 4.1.0)
## Rsamtools 2.8.0 2021-05-19 [1] Bioconductor
## RSpectra 0.16-0 2019-12-01 [1] RSPM (R 4.1.0)
## RSQLite 2.2.7 2021-04-22 [1] RSPM (R 4.1.0)
## rsvd 1.0.5 2021-04-16 [1] RSPM (R 4.1.0)
## rtracklayer 1.52.1 2021-08-15 [1] Bioconductor
## Rtsne 0.15 2018-11-10 [1] RSPM (R 4.1.0)
## S4Vectors * 0.30.0 2021-05-19 [1] Bioconductor
## sass 0.4.0 2021-05-12 [1] RSPM (R 4.1.0)
## ScaledMatrix 1.0.0 2021-05-19 [1] Bioconductor
## scales 1.1.1 2020-05-11 [1] RSPM (R 4.1.0)
## scater * 1.20.1 2021-06-15 [1] Bioconductor
## scPipe * 1.14.0 2021-05-19 [1] Bioconductor
## scran 1.20.1 2021-05-24 [1] Bioconductor
## scRNAseq * 2.6.1 2021-05-25 [1] Bioconductor
## scuttle * 1.2.1 2021-08-05 [1] Bioconductor
## sessioninfo 1.1.1 2018-11-05 [2] RSPM (R 4.1.0)
## shiny 1.6.0 2021-01-25 [1] RSPM (R 4.1.0)
## SingleCellExperiment * 1.14.1 2021-05-21 [1] Bioconductor
## sparseMatrixStats 1.4.2 2021-08-08 [1] Bioconductor
## statmod 1.4.36 2021-05-10 [1] RSPM (R 4.1.0)
## stringi 1.7.3 2021-07-16 [2] RSPM (R 4.1.0)
## stringr 1.4.0 2019-02-10 [2] RSPM (R 4.1.0)
## SummarizedExperiment * 1.22.0 2021-05-19 [1] Bioconductor
## tibble 3.1.3 2021-07-23 [2] RSPM (R 4.1.0)
## tidyselect 1.1.1 2021-04-30 [1] RSPM (R 4.1.0)
## utf8 1.2.2 2021-07-24 [2] RSPM (R 4.1.0)
## uwot 0.1.10 2020-12-15 [1] RSPM (R 4.1.0)
## vctrs 0.3.8 2021-04-29 [2] RSPM (R 4.1.0)
## vipor 0.4.5 2017-03-22 [1] RSPM (R 4.1.0)
## viridis 0.6.1 2021-05-11 [1] RSPM (R 4.1.0)
## viridisLite 0.4.0 2021-04-13 [1] RSPM (R 4.1.0)
## withr 2.4.2 2021-04-18 [2] RSPM (R 4.1.0)
## xfun 0.25 2021-08-06 [2] RSPM (R 4.1.0)
## XML 3.99-0.7 2021-08-17 [1] RSPM (R 4.1.0)
## xml2 1.3.2 2020-04-23 [2] RSPM (R 4.1.0)
## xtable 1.8-4 2019-04-21 [1] RSPM (R 4.1.0)
## XVector 0.32.0 2021-05-19 [1] Bioconductor
## yaml 2.2.1 2020-02-01 [2] RSPM (R 4.1.0)
## zlibbioc 1.38.0 2021-05-19 [1] Bioconductor
##
## [1] /__w/_temp/Library
## [2] /usr/local/lib/R/site-library
## [3] /usr/local/lib/R/libraryPatrocinadores
Agradecemos a nuestros patrocinadores:

