11 Anotación de clusters de células
Instructora: Yalbi I. Balderas-Martínez.
11.1 Diapositivas de Peter Hickey
Ver las diapositivas originales aquí
11.2 Motivación
Ahora estamos a punto de obtener la interpretación biológica de los resultados
Esta es la tarea más retadora en los análisis de datos scRNA-seq
👉 La obtención de clústeres es más o menos directa
🤔 ¿Cuál es el estado biológico que está representado por cada uno de los clústeres?
👉 Necesitamos hacer un puente entre el gap del dataset actual y el conocimiento biológico a priori (no siempre está disponible en una forma consistente y cualitativa)
🤔 ¿Qué es un tipo celular?
🔬 “Lo sabré cuando lo vea”
💻 “No”
Aplicaremos varios métodos computacionales que explotan la información a priori para asignar el significado a un dataset no caracterizado de scRNA-seq.
Algunas fuentes de información a priori
- Conjuntos de genes curados (e.g. Gene Ontology)
- Perfiles de expresión de bases de datos publicadas de referencia
- Los datos raros que tú hayas escondido en tu cerebro
11.3 Dataset ilustrativo: PBMC4k 10X sin filtrar
# Usemos datos de pbmc4k
library(BiocFileCache)
bfc <- BiocFileCache()
raw.path <- bfcrpath(bfc, file.path(
"http://cf.10xgenomics.com/samples",
"cell-exp/2.1.0/pbmc4k/pbmc4k_raw_gene_bc_matrices.tar.gz"
))
untar(raw.path, exdir = file.path(tempdir(), "pbmc4k"))
library(DropletUtils)
library(Matrix)
fname <- file.path(tempdir(), "pbmc4k/raw_gene_bc_matrices/GRCh38")
sce.pbmc <- read10xCounts(fname, col.names = TRUE)Dataset “Células mononucleares humanas de sangre periférica” de 10X Genomics
11.3.1 Anotación
# Anotación de los genes
library(scater)
rownames(sce.pbmc) <- uniquifyFeatureNames(
rowData(sce.pbmc)$ID, rowData(sce.pbmc)$Symbol
)
library(EnsDb.Hsapiens.v86)
location <- mapIds(EnsDb.Hsapiens.v86,
keys = rowData(sce.pbmc)$ID,
column = "SEQNAME", keytype = "GENEID"
)
# Detección de _droplets_ con células
set.seed(100)
e.out <- emptyDrops(counts(sce.pbmc))
sce.pbmc <- sce.pbmc[, which(e.out$FDR <= 0.001)]11.3.2 Control de calidad
# Control de calidad
stats <- perCellQCMetrics(sce.pbmc,
subsets = list(Mito = which(location == "MT"))
)
high.mito <- isOutlier(stats$subsets_Mito_percent,
type = "higher"
)
sce.pbmc <- sce.pbmc[, !high.mito]
# Normalización de los datos
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.pbmc)
sce.pbmc <- computeSumFactors(sce.pbmc, cluster = clusters)
sce.pbmc <- logNormCounts(sce.pbmc)11.3.3 Genes variables
## Identificación de genes altamente variables
set.seed(1001)
dec.pbmc <- modelGeneVarByPoisson(sce.pbmc)
top.pbmc <- getTopHVGs(dec.pbmc, prop = 0.1)11.3.4 Reducción de dimensiones
## Reducción de dimensiones
set.seed(10000)
sce.pbmc <- denoisePCA(sce.pbmc,
subset.row = top.pbmc,
technical = dec.pbmc
)
set.seed(100000)
sce.pbmc <- runTSNE(sce.pbmc, dimred = "PCA")
set.seed(1000000)
sce.pbmc <- runUMAP(sce.pbmc, dimred = "PCA")11.3.5 Clustering
# clustering
g <- buildSNNGraph(sce.pbmc, k = 10, use.dimred = "PCA")
clust <- igraph::cluster_walktrap(g)$membership
sce.pbmc$cluster <- factor(clust)11.4 Asignando las etiquetas celulares a partir de los datos de referencia
11.4.1 Visión general
👉 Un enfoque directo es comparar los perfiles de expresión single-cell con datasets previamente anotados
👉 Las etiquetas pueden entonces ser asignadas a cada célula en nuestro dataset no caracterizado de prueba basado en la muestra de referencia más similar, por dar alguna definición de “similar”
👉 Cualquier dataset de expresión génica etiquetado (microarreglos, RNA-seq bulk, scRNA-seq) puede ser usado como una referencia
⚠️ Sin embargo, su confiabilidad depende enormemente en la calidad de los datos originales y la experiencia de los autores originales quienes asignaron las etiquetas en primer lugar
👉 Asignar las etiquetas a un dataset de “prueba” a partir de un dataset de “entrenamiento” (referencia), es un problema estándar en estadística / machine learning
👉 Usaremos el método SingleR (Aran et al. 2019)
11.5 SingleR
- 🤓 Asigna las etiquetas a las células basado en las muestras de referencia con las correlaciones de rangos más altas de Spearman
- 🤓 Para reducir el ruido, identifica genes marcadores entre pares de etiquetas (en la referencia) y calcula la correlación usando solamente esos marcadores
- 🤓 Hace algún tipo de tuneado fino, repitiendo las correlaciones solamente con los genes marcadores de las etiquetas con el mejor score, ayudando a resolver cualquier ambigüedad entre esas etiquetas al eliminar el ruido a partir de marcadores irrelevantes para otras etiquetas
11.5.1 SingleR incluye varias referencias
# Human
celldex::BlueprintEncodeData()
celldex::DatabaseImmuneCellExpressionData()
celldex::HumanPrimaryCellAtlasData()
celldex::MonacoImmuneData()
celldex::NovershternHematopoieticData()
# Mice
celldex::ImmGenData()
celldex::MouseRNASeqData()11.5.2 Usando las referencias
# if needed install celldex
# create directory? y
library(celldex)
ref <- celldex::BlueprintEncodeData()❓ ¿Qué tipos celulares están disponibles en este dataset de referencia?
11.5.3 Usando las referencias integradas
library(SingleR)
pred <- SingleR(
test = sce.pbmc, ref = ref,
labels = ref$label.main
)- ❓ ¿Qué etiquetas han sido asignadas a los datos single-cell?
- ❓ ¿Cómo usaríamos las etiquetas “finas” con SingleR?
plotScoreHeatmap(pred)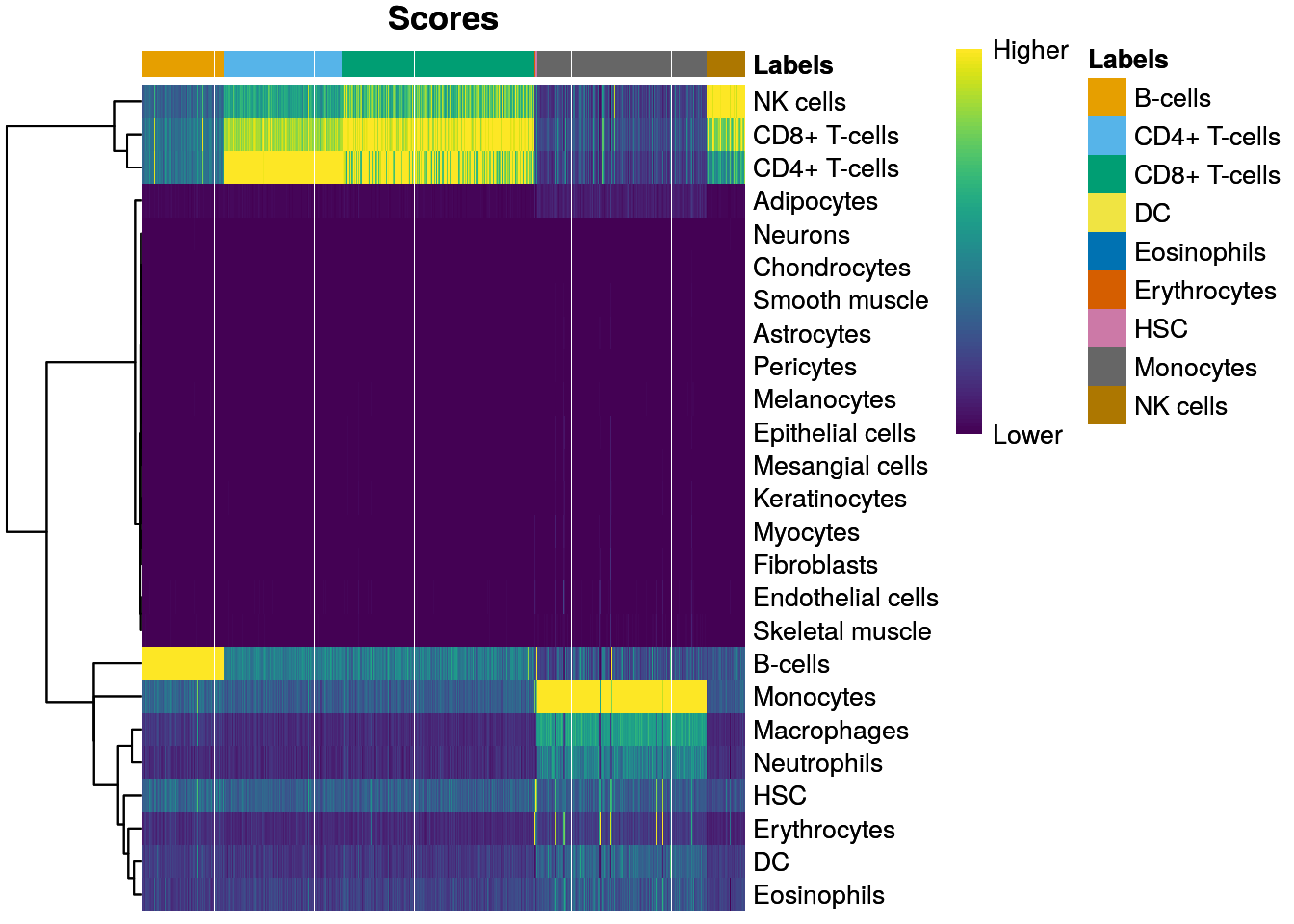
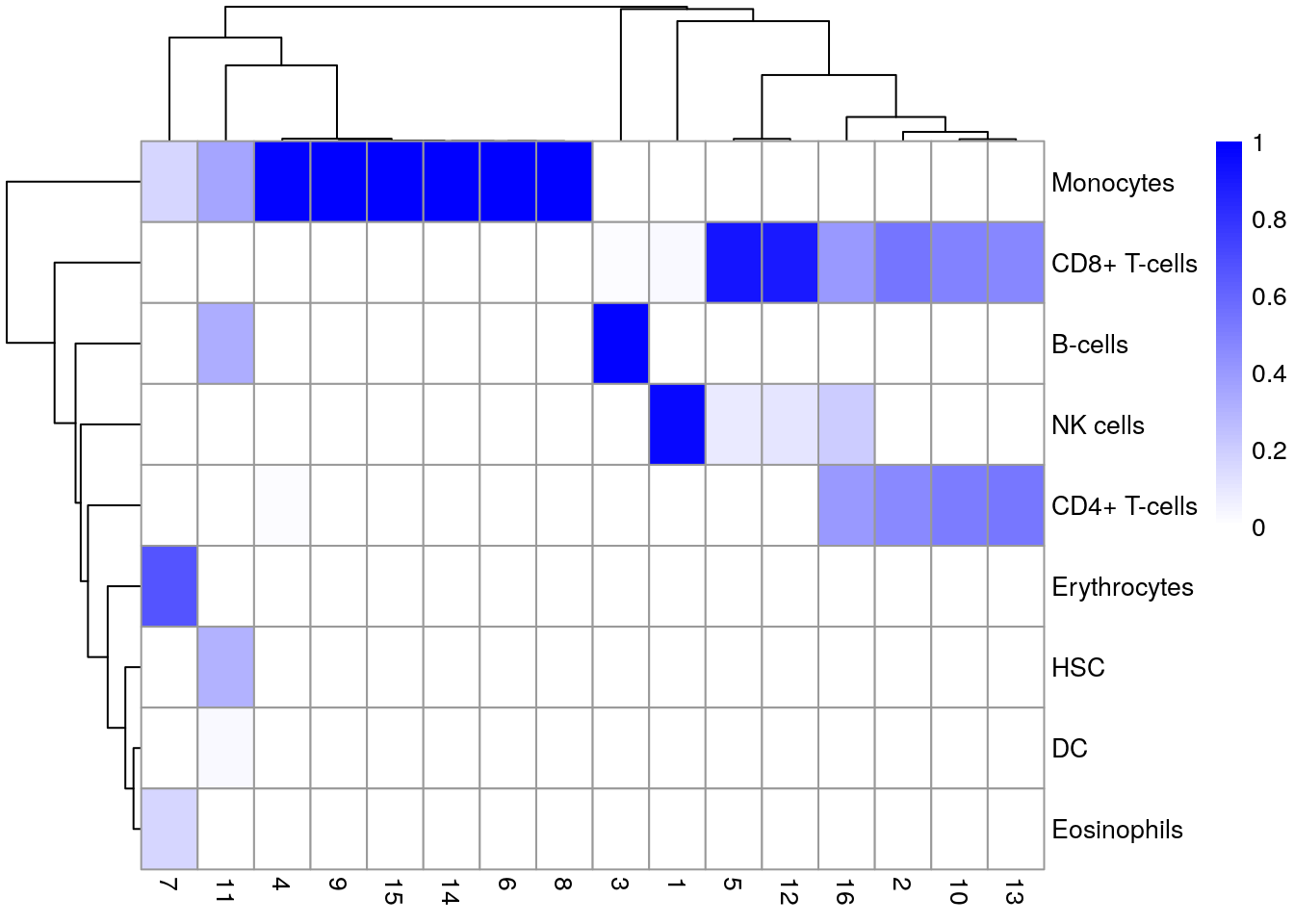
- 👉 Inspeccionamos los resultados usando un heatmap de los scores por célula y por etiqueta
- 👉 Idealmente, cada célula debería exhibir un score alto en una etiqueta relativa a todas las otras
- 👉 Los scores se muestran antes de cualquier tuneado fino y son normalizadas a [0, 1] dentro de cada célula
11.5.4 Podado de etiquetas (Label pruning)
total_pruned <- sum(is.na(pred$pruned.labels))
plotScoreHeatmap(pred, show.pruned = TRUE)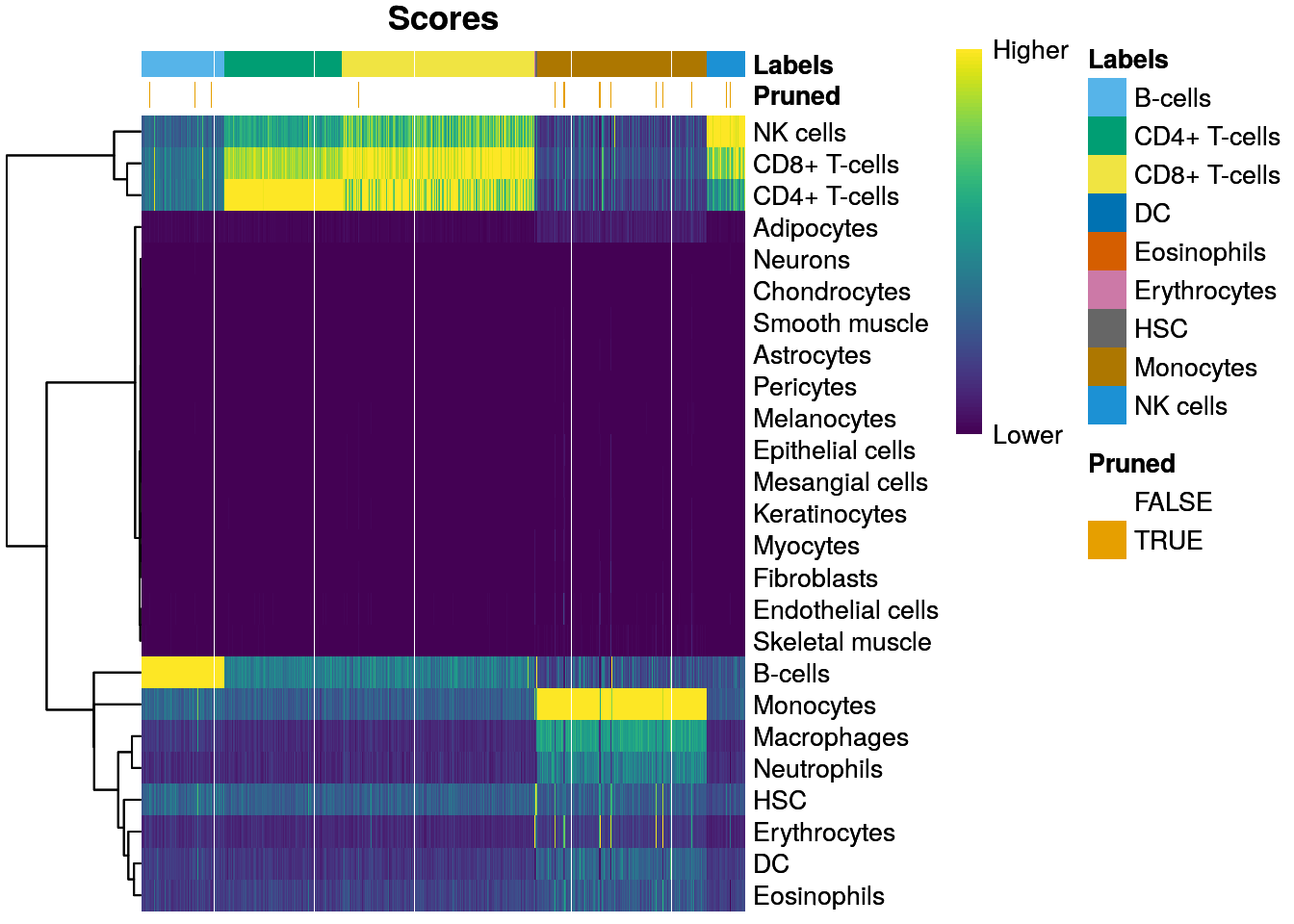
- 👉 SingleR intentará podar aquellas asignaciones de baja calidad marcándolas como NA
- 🤓 El podado se hace calculando la diferencia del score de la etiqueta asignada a partir del score de la mediana dentro de cada célula y entonces podando las células con un valor pequeño de esta diferencia
plotScoreDistribution(pred)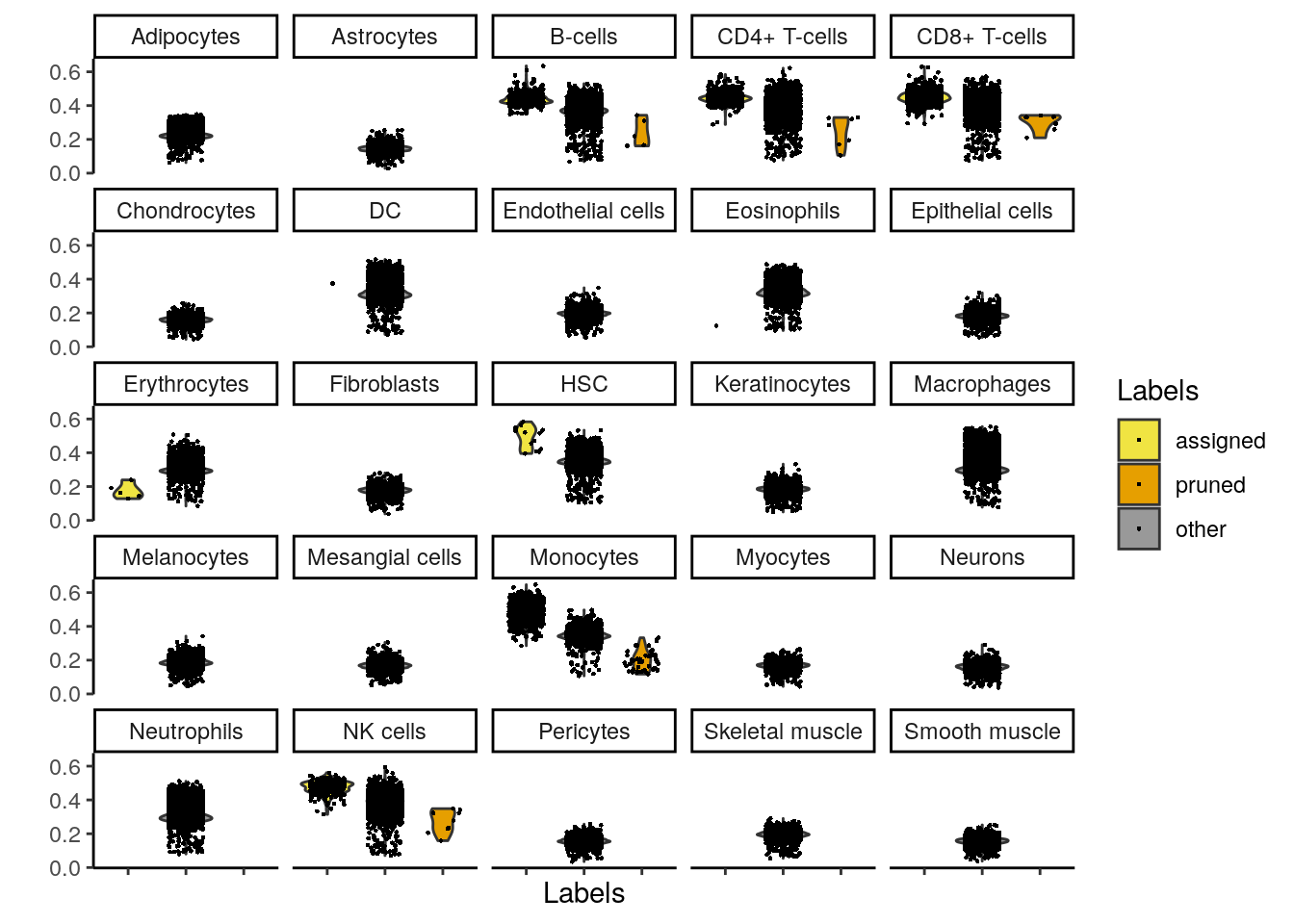
👉 Distribución de las diferencias del score de la etiqueta asignada a partir del score de la mediana dentro de cada célula
11.5.5 Identificando los genes con anotación dirigida
- 🤔 ¿Por qué las células en este clúster se etiquetan como el tipo celular X?
- 👉 Examina la expresión de los genes marcadores para cada etiqueta en el dataset de prueba
- 👉 Si una célula en el dataset de prueba está asignado con confianza a una etiqueta en particular, uno esperaría que tenga una fuerte expresión de los marcadores de esa etiqueta (al menos sobreexpresión con respecto a las células asignadas a otras etiquetas)
# install gmp, ClusterR, mbkmeans dependencies if needed
sce.pbmc$labels <- pred$labels
all.markers <- metadata(pred)$de.genes
lab <- "B-cells"
# Get top-10 marker genes for B-cells compared to each other cell
# type
top.markers <- Reduce(union, sapply(all.markers[[lab]], head, 10))
plotHeatmap(sce.pbmc,
order_columns_by = "labels",
features = top.markers, center = TRUE, zlim = c(-3, 3), main = lab
)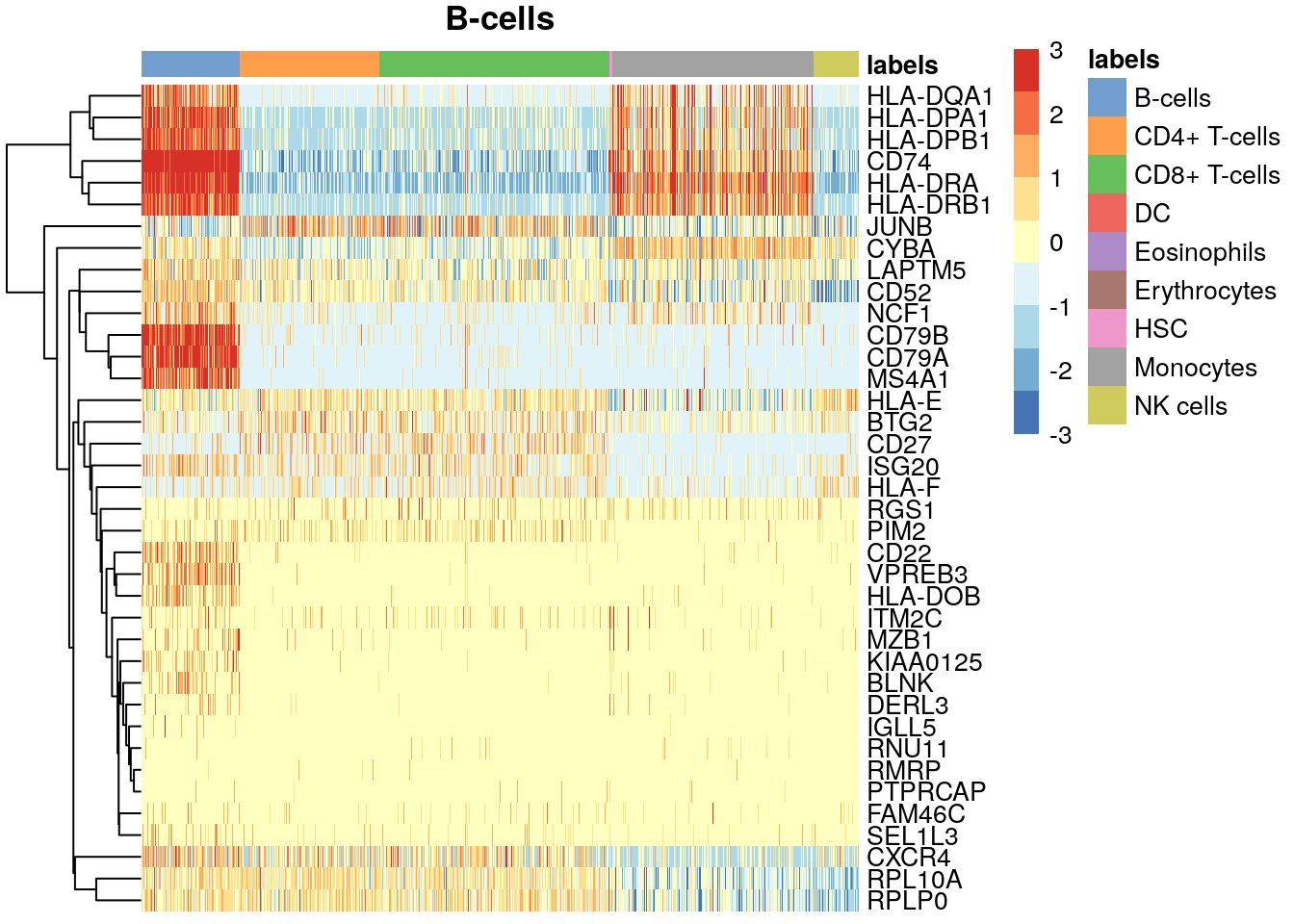
❓ Toma otro tipo celular e identifica los genes que dirigen la anotación
11.5.6 Comparando las etiquetas con los clústeres
tab <- table(Assigned = pred$pruned.labels, Cluster = sce.pbmc$cluster)
library(pheatmap)
# Proportion of cells in each cluster assigned to each label
pheatmap(prop.table(tab, margin = 2),
color = colorRampPalette(c("white", "blue"))(101)
)
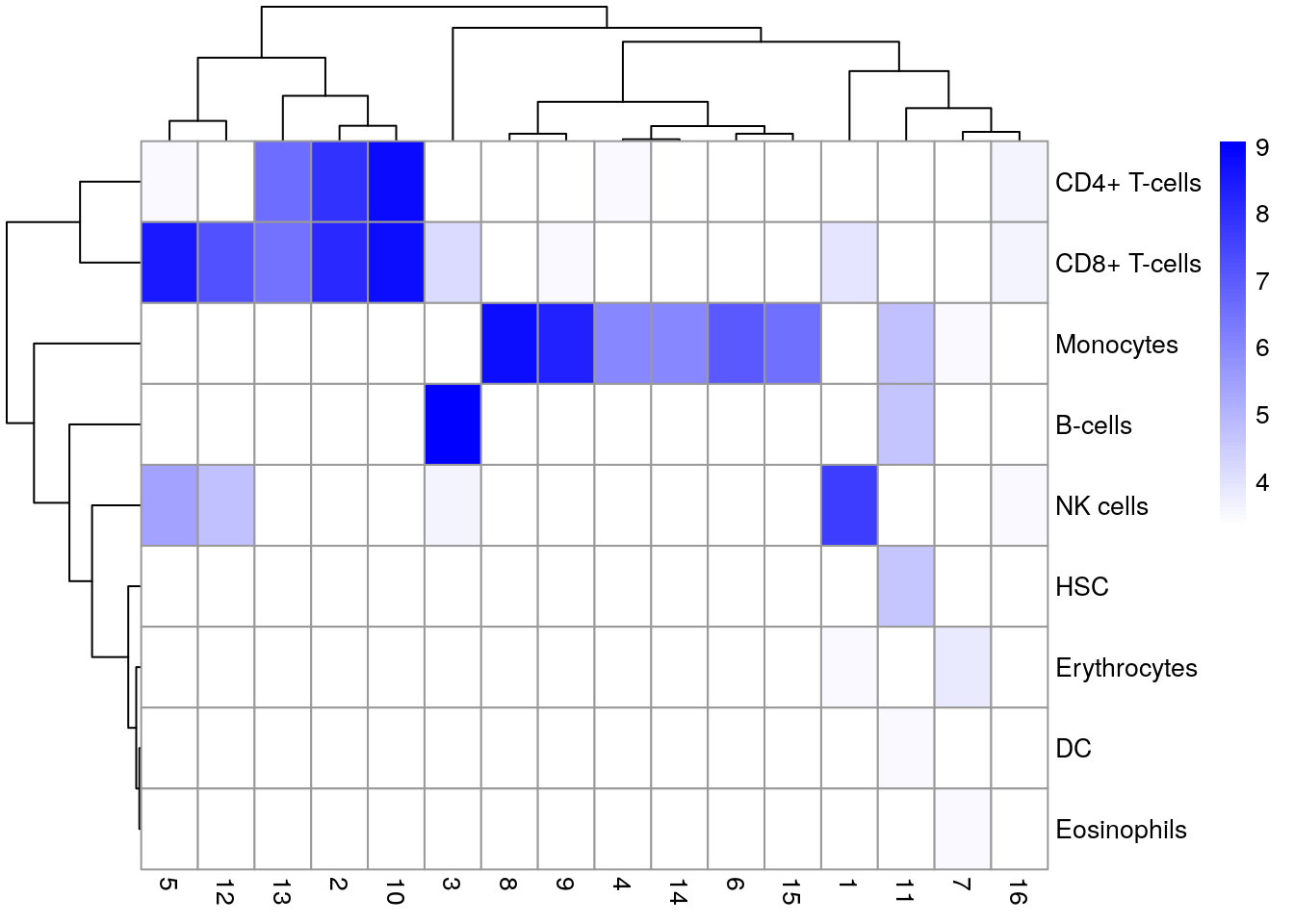
# (log-)number of cells in each cluster assigned to each label
# Adding a pseudo-count of 10 to avoid strong color jumps with just
# 1 cell.
pheatmap(log2(tab + 10),
color = colorRampPalette(c("white", "blue"))(101)
)
11.5.7 Voilà
plotTSNE(sce.pbmc, colour_by = "labels", text_by = "labels")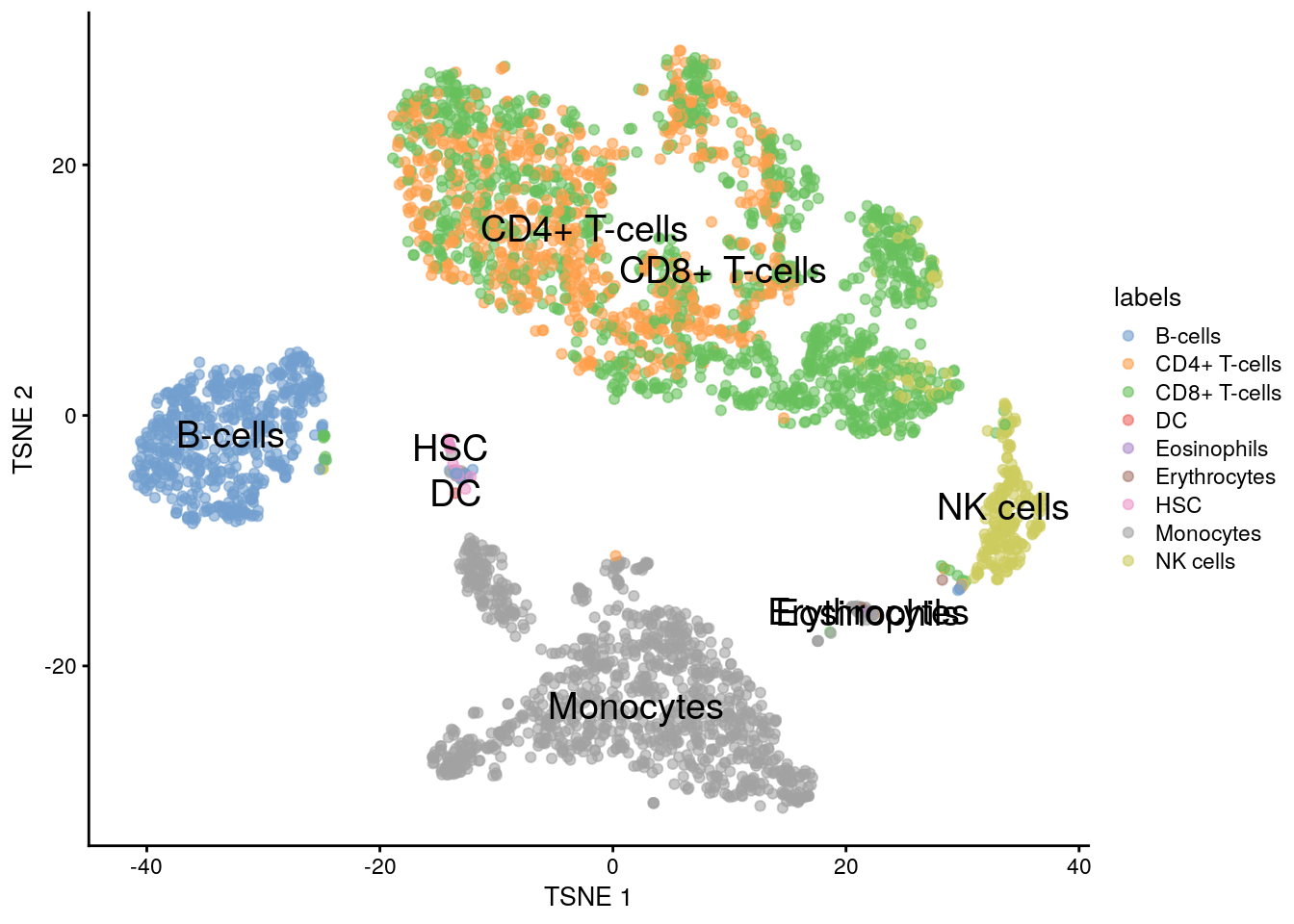
plotTSNE(sce.pbmc, colour_by = "cluster", text_by = "labels")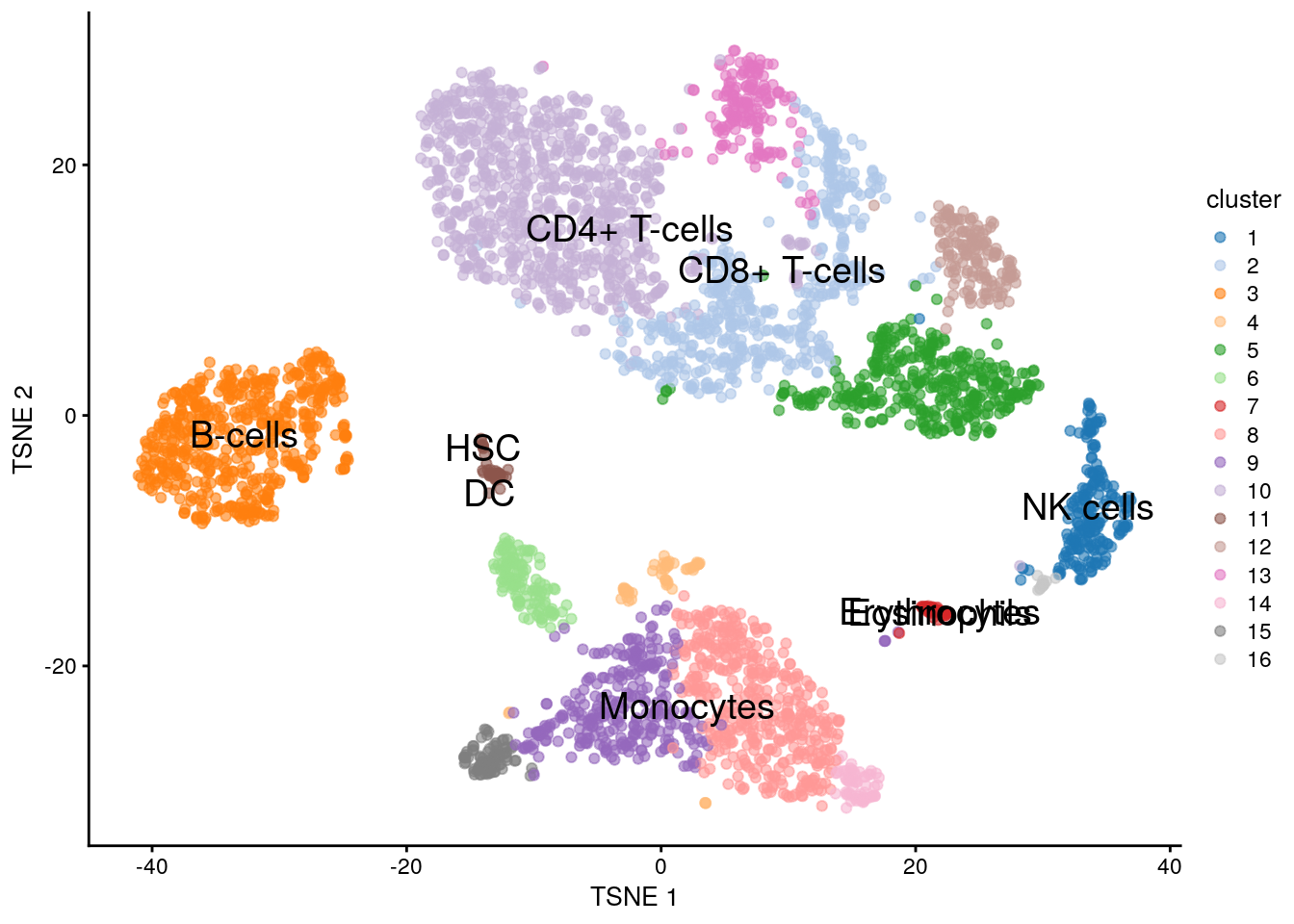
11.5.7.1 Aventura en el tiempo
Hmm. We can't figure out why these two plots are different with the same code, though different R versions.
— 🇲🇽 Leonardo Collado-Torres (@lcolladotor) August 10, 2021
BioC 3.11? @PeteHaitch https://t.co/8DJM6pagdj
BioC 3.13https://t.co/t8KiPTyU9D@Bioconductor #rstats @yalbi_ibm @AnaBetty2304 pic.twitter.com/P65u7dnVCo
Ok, not identical, but not bad with BioC 3.13 only
— 🇲🇽 Leonardo Collado-Torres (@lcolladotor) August 11, 2021
Left: BioC 3.13 with e.out from BioC 3.111
Middle: Pete's slides with BioC 3.11
Right: BioC 3.13 only#rstats #OSCA @Bioconductor #DropletUtils pic.twitter.com/d41LiuXKWn
11.6 Resumen de la anotación basada en una referencia (e.g., SingleR)
- ➕ Se centra en aspectos de los datos que se sabe son interesantes, simplifica el proceso de la interpretación biológica
- ➖ Está restringido por la diversidad y la resolución de las etiquetas disponibles en el dataset de referencia
- 👉 Se pueden suplir referencias personalizadas a SingleR
11.7 Asignando las etiquetas de tipos celulares a partir de marcadores
- 🤔 ¿Cómo podemos hacer uso de nuestros genes marcadores agrupados?
- 🥉 Revisarlos en hojas de cálculo
- 🥈 Observar heatmaps
- 🥇 Realizar un gene set enrichment analysis
11.7.1 Gene set enrichment analysis
- 👉 Identifica las rutas y procesos que están (relativamente) activos en cada clúster basado en la sobreexpresión de los genes asociados en comparación con otros clústeres
- ➕ Un método confiable para determinar si las rutas están sobre- o sub- expresadas entre clúesteres
- ➕ Existen un montón de herramientas para gene set enrichment analysis
- ➖ Todas las conclusiones son relativas a otros clústeres, haciéndolo más difícil para determinar la identidad celular si alguno no está presente en el mismo estudio más info
11.7.2 Calculando las actividades de los conjuntos de genes
- 👉 Calcular el promedio de la expresión en log en todos los genes, en un conjunto de genes para cada célula y examinar los clústeres con valores altos (gene set activities)
- 👉 Se necesita proveer de conjuntos de genes
- ➖ No todos los genes en el conjunto pueden exhibir el mismo patrón de diferencia y los genes no-DE añadirán ruido, “diluyendo” la fuerza de cualquiera de las diferencias comparadas a un análisis que se centra directamente en genes DE
- 👉 Es más una visualización útil que la base para cualquier análisis estadístico real más info
11.8 Resumen y recomendaciones
- 👉 La anotación de tipos celulares “automática”, como SingleR, es mejor cuando funciona (i.e. cuando hay un dataset de referencia apropiado)
- 👉 Usualmente necesitaremos usar un método manual, como aquellos basados en agrupar los genes marcadores (e.g., gene set enrichment analysis)
- 👉 La anotación del tipo celular ofrecerá una reconsideración inmediata de los parámetros del agrupamiento y/o algunos retoques manuales a los clústeres
11.9 Detalles de la sesión de R
## Información de la sesión de R
Sys.time()## [1] "2021-08-19 15:42:29 UTC"proc.time()## user system elapsed
## 200.819 4.594 202.975options(width = 120)
sessioninfo::session_info()## ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
## setting value
## version R version 4.1.0 (2021-05-18)
## os Ubuntu 20.04.2 LTS
## system x86_64, linux-gnu
## ui X11
## language (EN)
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz UTC
## date 2021-08-19
##
## ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
## package * version date lib source
## AnnotationDbi * 1.54.1 2021-06-08 [1] Bioconductor
## AnnotationFilter * 1.16.0 2021-05-19 [1] Bioconductor
## AnnotationHub 3.0.1 2021-06-20 [1] Bioconductor
## assertthat 0.2.1 2019-03-21 [1] RSPM (R 4.1.0)
## beachmat 2.8.1 2021-08-12 [1] Bioconductor
## beeswarm 0.4.0 2021-06-01 [1] RSPM (R 4.1.0)
## Biobase * 2.52.0 2021-05-19 [1] Bioconductor
## BiocFileCache * 2.0.0 2021-05-19 [1] Bioconductor
## BiocGenerics * 0.38.0 2021-05-19 [1] Bioconductor
## BiocIO 1.2.0 2021-05-19 [1] Bioconductor
## BiocManager 1.30.16 2021-06-15 [1] RSPM (R 4.1.0)
## BiocNeighbors 1.10.0 2021-05-19 [1] Bioconductor
## BiocParallel 1.26.1 2021-07-04 [1] Bioconductor
## BiocSingular 1.8.1 2021-06-08 [1] Bioconductor
## BiocVersion 3.13.1 2021-03-19 [2] Bioconductor
## biomaRt 2.48.3 2021-08-15 [1] Bioconductor
## Biostrings 2.60.2 2021-08-05 [1] Bioconductor
## bit 4.0.4 2020-08-04 [1] RSPM (R 4.1.0)
## bit64 4.0.5 2020-08-30 [1] RSPM (R 4.1.0)
## bitops 1.0-7 2021-04-24 [1] RSPM (R 4.1.0)
## blob 1.2.2 2021-07-23 [1] RSPM (R 4.1.0)
## bluster 1.2.1 2021-05-27 [1] Bioconductor
## bookdown 0.23 2021-08-13 [1] RSPM (R 4.1.0)
## bslib 0.2.5.1 2021-05-18 [1] RSPM (R 4.1.0)
## cachem 1.0.5 2021-05-15 [2] RSPM (R 4.1.0)
## celldex * 1.2.0 2021-05-20 [1] Bioconductor
## cli 3.0.1 2021-07-17 [2] RSPM (R 4.1.0)
## cluster 2.1.2 2021-04-17 [3] CRAN (R 4.1.0)
## colorspace 2.0-2 2021-06-24 [1] RSPM (R 4.1.0)
## cowplot 1.1.1 2020-12-30 [1] RSPM (R 4.1.0)
## crayon 1.4.1 2021-02-08 [2] RSPM (R 4.1.0)
## curl 4.3.2 2021-06-23 [2] RSPM (R 4.1.0)
## DBI 1.1.1 2021-01-15 [1] RSPM (R 4.1.0)
## dbplyr * 2.1.1 2021-04-06 [1] RSPM (R 4.1.0)
## DelayedArray 0.18.0 2021-05-19 [1] Bioconductor
## DelayedMatrixStats 1.14.2 2021-08-08 [1] Bioconductor
## digest 0.6.27 2020-10-24 [2] RSPM (R 4.1.0)
## dplyr 1.0.7 2021-06-18 [1] RSPM (R 4.1.0)
## dqrng 0.3.0 2021-05-01 [1] RSPM (R 4.1.0)
## DropletUtils * 1.12.2 2021-07-22 [1] Bioconductor
## edgeR 3.34.0 2021-05-19 [1] Bioconductor
## ellipsis 0.3.2 2021-04-29 [2] RSPM (R 4.1.0)
## EnsDb.Hsapiens.v86 * 2.99.0 2021-07-29 [1] Bioconductor
## ensembldb * 2.16.4 2021-08-05 [1] Bioconductor
## evaluate 0.14 2019-05-28 [2] RSPM (R 4.1.0)
## ExperimentHub 2.0.0 2021-05-19 [1] Bioconductor
## fansi 0.5.0 2021-05-25 [2] RSPM (R 4.1.0)
## farver 2.1.0 2021-02-28 [1] RSPM (R 4.1.0)
## fastmap 1.1.0 2021-01-25 [2] RSPM (R 4.1.0)
## filelock 1.0.2 2018-10-05 [1] RSPM (R 4.1.0)
## FNN 1.1.3 2019-02-15 [1] RSPM (R 4.1.0)
## generics 0.1.0 2020-10-31 [1] RSPM (R 4.1.0)
## GenomeInfoDb * 1.28.1 2021-07-01 [1] Bioconductor
## GenomeInfoDbData 1.2.6 2021-07-29 [1] Bioconductor
## GenomicAlignments 1.28.0 2021-05-19 [1] Bioconductor
## GenomicFeatures * 1.44.1 2021-08-15 [1] Bioconductor
## GenomicRanges * 1.44.0 2021-05-19 [1] Bioconductor
## ggbeeswarm 0.6.0 2017-08-07 [1] RSPM (R 4.1.0)
## ggplot2 * 3.3.5 2021-06-25 [1] RSPM (R 4.1.0)
## glue 1.4.2 2020-08-27 [2] RSPM (R 4.1.0)
## gridExtra 2.3 2017-09-09 [1] RSPM (R 4.1.0)
## gtable 0.3.0 2019-03-25 [1] RSPM (R 4.1.0)
## HDF5Array 1.20.0 2021-05-19 [1] Bioconductor
## highr 0.9 2021-04-16 [2] RSPM (R 4.1.0)
## hms 1.1.0 2021-05-17 [1] RSPM (R 4.1.0)
## htmltools 0.5.1.1 2021-01-22 [1] RSPM (R 4.1.0)
## httpuv 1.6.1 2021-05-07 [1] RSPM (R 4.1.0)
## httr 1.4.2 2020-07-20 [2] RSPM (R 4.1.0)
## igraph 1.2.6 2020-10-06 [1] RSPM (R 4.1.0)
## interactiveDisplayBase 1.30.0 2021-05-19 [1] Bioconductor
## IRanges * 2.26.0 2021-05-19 [1] Bioconductor
## irlba 2.3.3 2019-02-05 [1] RSPM (R 4.1.0)
## jquerylib 0.1.4 2021-04-26 [1] RSPM (R 4.1.0)
## jsonlite 1.7.2 2020-12-09 [2] RSPM (R 4.1.0)
## KEGGREST 1.32.0 2021-05-19 [1] Bioconductor
## knitr 1.33 2021-04-24 [2] RSPM (R 4.1.0)
## labeling 0.4.2 2020-10-20 [1] RSPM (R 4.1.0)
## later 1.2.0 2021-04-23 [1] RSPM (R 4.1.0)
## lattice 0.20-44 2021-05-02 [3] CRAN (R 4.1.0)
## lazyeval 0.2.2 2019-03-15 [1] RSPM (R 4.1.0)
## lifecycle 1.0.0 2021-02-15 [2] RSPM (R 4.1.0)
## limma 3.48.3 2021-08-10 [1] Bioconductor
## locfit 1.5-9.4 2020-03-25 [1] RSPM (R 4.1.0)
## magrittr 2.0.1 2020-11-17 [2] RSPM (R 4.1.0)
## Matrix * 1.3-4 2021-06-01 [3] RSPM (R 4.1.0)
## MatrixGenerics * 1.4.2 2021-08-08 [1] Bioconductor
## matrixStats * 0.60.0 2021-07-26 [1] RSPM (R 4.1.0)
## memoise 2.0.0 2021-01-26 [2] RSPM (R 4.1.0)
## metapod 1.0.0 2021-05-19 [1] Bioconductor
## mime 0.11 2021-06-23 [2] RSPM (R 4.1.0)
## munsell 0.5.0 2018-06-12 [1] RSPM (R 4.1.0)
## pheatmap * 1.0.12 2019-01-04 [1] RSPM (R 4.1.0)
## pillar 1.6.2 2021-07-29 [2] RSPM (R 4.1.0)
## pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.1.0)
## png 0.1-7 2013-12-03 [1] RSPM (R 4.1.0)
## prettyunits 1.1.1 2020-01-24 [2] RSPM (R 4.1.0)
## progress 1.2.2 2019-05-16 [1] RSPM (R 4.1.0)
## promises 1.2.0.1 2021-02-11 [1] RSPM (R 4.1.0)
## ProtGenerics 1.24.0 2021-05-19 [1] Bioconductor
## purrr 0.3.4 2020-04-17 [2] RSPM (R 4.1.0)
## R.methodsS3 1.8.1 2020-08-26 [1] RSPM (R 4.1.0)
## R.oo 1.24.0 2020-08-26 [1] RSPM (R 4.1.0)
## R.utils 2.10.1 2020-08-26 [1] RSPM (R 4.1.0)
## R6 2.5.0 2020-10-28 [2] RSPM (R 4.1.0)
## rappdirs 0.3.3 2021-01-31 [2] RSPM (R 4.1.0)
## RColorBrewer 1.1-2 2014-12-07 [1] RSPM (R 4.1.0)
## Rcpp 1.0.7 2021-07-07 [2] RSPM (R 4.1.0)
## RCurl 1.98-1.4 2021-08-17 [1] RSPM (R 4.1.0)
## restfulr 0.0.13 2017-08-06 [1] RSPM (R 4.1.0)
## rhdf5 2.36.0 2021-05-19 [1] Bioconductor
## rhdf5filters 1.4.0 2021-05-19 [1] Bioconductor
## Rhdf5lib 1.14.2 2021-07-06 [1] Bioconductor
## rjson 0.2.20 2018-06-08 [1] RSPM (R 4.1.0)
## rlang 0.4.11 2021-04-30 [2] RSPM (R 4.1.0)
## rmarkdown 2.10 2021-08-06 [1] RSPM (R 4.1.0)
## Rsamtools 2.8.0 2021-05-19 [1] Bioconductor
## RSpectra 0.16-0 2019-12-01 [1] RSPM (R 4.1.0)
## RSQLite 2.2.7 2021-04-22 [1] RSPM (R 4.1.0)
## rsvd 1.0.5 2021-04-16 [1] RSPM (R 4.1.0)
## rtracklayer 1.52.1 2021-08-15 [1] Bioconductor
## Rtsne 0.15 2018-11-10 [1] RSPM (R 4.1.0)
## S4Vectors * 0.30.0 2021-05-19 [1] Bioconductor
## sass 0.4.0 2021-05-12 [1] RSPM (R 4.1.0)
## ScaledMatrix 1.0.0 2021-05-19 [1] Bioconductor
## scales 1.1.1 2020-05-11 [1] RSPM (R 4.1.0)
## scater * 1.20.1 2021-06-15 [1] Bioconductor
## scran * 1.20.1 2021-05-24 [1] Bioconductor
## scuttle * 1.2.1 2021-08-05 [1] Bioconductor
## sessioninfo 1.1.1 2018-11-05 [2] RSPM (R 4.1.0)
## shiny 1.6.0 2021-01-25 [1] RSPM (R 4.1.0)
## SingleCellExperiment * 1.14.1 2021-05-21 [1] Bioconductor
## SingleR * 1.6.1 2021-05-20 [1] Bioconductor
## sparseMatrixStats 1.4.2 2021-08-08 [1] Bioconductor
## statmod 1.4.36 2021-05-10 [1] RSPM (R 4.1.0)
## stringi 1.7.3 2021-07-16 [2] RSPM (R 4.1.0)
## stringr 1.4.0 2019-02-10 [2] RSPM (R 4.1.0)
## SummarizedExperiment * 1.22.0 2021-05-19 [1] Bioconductor
## tibble 3.1.3 2021-07-23 [2] RSPM (R 4.1.0)
## tidyselect 1.1.1 2021-04-30 [1] RSPM (R 4.1.0)
## utf8 1.2.2 2021-07-24 [2] RSPM (R 4.1.0)
## uwot 0.1.10 2020-12-15 [1] RSPM (R 4.1.0)
## vctrs 0.3.8 2021-04-29 [2] RSPM (R 4.1.0)
## vipor 0.4.5 2017-03-22 [1] RSPM (R 4.1.0)
## viridis 0.6.1 2021-05-11 [1] RSPM (R 4.1.0)
## viridisLite 0.4.0 2021-04-13 [1] RSPM (R 4.1.0)
## withr 2.4.2 2021-04-18 [2] RSPM (R 4.1.0)
## xfun 0.25 2021-08-06 [2] RSPM (R 4.1.0)
## XML 3.99-0.7 2021-08-17 [1] RSPM (R 4.1.0)
## xml2 1.3.2 2020-04-23 [2] RSPM (R 4.1.0)
## xtable 1.8-4 2019-04-21 [1] RSPM (R 4.1.0)
## XVector 0.32.0 2021-05-19 [1] Bioconductor
## yaml 2.2.1 2020-02-01 [2] RSPM (R 4.1.0)
## zlibbioc 1.38.0 2021-05-19 [1] Bioconductor
##
## [1] /__w/_temp/Library
## [2] /usr/local/lib/R/site-library
## [3] /usr/local/lib/R/libraryPatrocinadores
Agradecemos a nuestros patrocinadores:


Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).↩︎