class: center, middle, inverse, title-slide # Minicurso ### Ana BVA, Rodolfo LCD ### 15 April 2021 --- class: inverse, center, middle # Agradecimientos <style type="text/css"> /* From https://github.com/yihui/xaringan/issues/147 */ .scroll-output { height: 80%; overflow-y: scroll; } /* https://stackoverflow.com/questions/50919104/horizontally-scrollable-output-on-xaringan-slides */ pre { max-width: 100%; overflow-x: scroll; } .remark-slide-content { font-size: 28px; padding: 20px 80px 20px 80px; } .remark-code, .remark-inline-code { background: #f0f0f0; } .remark-code { font-size: 24px; } .huge .remark-code { /*Change made here*/ font-size: 200% !important; } .tiny .remark-code { /*Change made here*/ font-size: 80% !important; } .center2 { margin: 0; position: absolute; top: 50%; left: 50%; -ms-transform: translate(-50%, -50%); transform: translate(-50%, -50%); } </style> --- ## Agradecimientos Este material está basado en [Love, Michael I., et al. "RNA-Seq workflow: gene-level exploratory analysis and differential expression." F1000Research 4 (2015).](https://www.bioconductor.org/packages/devel/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html) de Bioconductor. <img src="https://images.squarespace-cdn.com/content/v1/5423875be4b03f0c482a58c4/1532953317705-W6TYTE70KG5E6KQEZU1K/ke17ZwdGBToddI8pDm48kNVP8RwsgCc7XlHc2zPQeqdZw-zPPgdn4jUwVcJE1ZvWQUxwkmyExglNqGp0IvTJZUJFbgE-7XRK3dMEBRBhUpxA5wn4368HhrDKfvXqORu9oTEmTJkjlQ2gdQcVngofpWQcE5w-MKnvigIPwIMqpXs/Bioconductor.png" width="300px" style="display: block; margin: auto;" /> --- ## Agradecimientos También está basado en el curso de ["Next generation sequencing bioiformatics" (Santiago, Chile 2019)](https://coursesandconferences.wellcomegenomecampus.org/our-events/next-generation-sequencing-bioinformatics-chile-2019/). <img src="./Images/module7.png" width="600px" style="display: block; margin: auto;" /> --- ## Agradecimientos <img src="https://i0.wp.com/alcanzandoelconocimiento.com/wp-content/uploads/2019/11/Conabio-Logo-02.jpg?w=1024&ssl=1" width="600px" style="display: block; margin: auto;" /> --- class: inverse, center, middle # 1: Introducción al RNA-seq --- ## Transcriptómica Estudio de la expresión genética - ¿Cuánto RNA hay? <img src="./Images/transcriptomica.png" width="900px" style="display: block; margin: auto;" /> --- ## Tecnologías <img src="./Images/book.png" width="800px" style="display: block; margin: auto;" /> --- ## RNA-seq <img src="https://upload.wikimedia.org/wikipedia/commons/thumb/f/f3/Summary_of_RNA-Seq.svg/800px-Summary_of_RNA-Seq.svg.png" width="660px" style="display: block; margin: auto;" /> --- ## Aplicaciones <img src="./Images/book.png" width="570px" style="display: block; margin: auto;" /> --- ## Bases de datos <img src="./Images/book.png" width="450px" style="display: block; margin: auto;" /> --- class: inverse, center, middle # 2: Organización de un proyecto bioinformático --- ## Organización del proyecto <img src="./Images/Repo.png" width="400px" style="display: block; margin: auto;" /> Almacenar el proyecto en .blue[GitHub] <svg viewBox="0 0 496 512" style="height:1em;position:relative;display:inline-block;top:.1em;fill:blue;" xmlns="http://www.w3.org/2000/svg"> <path d="M165.9 397.4c0 2-2.3 3.6-5.2 3.6-3.3.3-5.6-1.3-5.6-3.6 0-2 2.3-3.6 5.2-3.6 3-.3 5.6 1.3 5.6 3.6zm-31.1-4.5c-.7 2 1.3 4.3 4.3 4.9 2.6 1 5.6 0 6.2-2s-1.3-4.3-4.3-5.2c-2.6-.7-5.5.3-6.2 2.3zm44.2-1.7c-2.9.7-4.9 2.6-4.6 4.9.3 2 2.9 3.3 5.9 2.6 2.9-.7 4.9-2.6 4.6-4.6-.3-1.9-3-3.2-5.9-2.9zM244.8 8C106.1 8 0 113.3 0 252c0 110.9 69.8 205.8 169.5 239.2 12.8 2.3 17.3-5.6 17.3-12.1 0-6.2-.3-40.4-.3-61.4 0 0-70 15-84.7-29.8 0 0-11.4-29.1-27.8-36.6 0 0-22.9-15.7 1.6-15.4 0 0 24.9 2 38.6 25.8 21.9 38.6 58.6 27.5 72.9 20.9 2.3-16 8.8-27.1 16-33.7-55.9-6.2-112.3-14.3-112.3-110.5 0-27.5 7.6-41.3 23.6-58.9-2.6-6.5-11.1-33.3 2.6-67.9 20.9-6.5 69 27 69 27 20-5.6 41.5-8.5 62.8-8.5s42.8 2.9 62.8 8.5c0 0 48.1-33.6 69-27 13.7 34.7 5.2 61.4 2.6 67.9 16 17.7 25.8 31.5 25.8 58.9 0 96.5-58.9 104.2-114.8 110.5 9.2 7.9 17 22.9 17 46.4 0 33.7-.3 75.4-.3 83.6 0 6.5 4.6 14.4 17.3 12.1C428.2 457.8 496 362.9 496 252 496 113.3 383.5 8 244.8 8zM97.2 352.9c-1.3 1-1 3.3.7 5.2 1.6 1.6 3.9 2.3 5.2 1 1.3-1 1-3.3-.7-5.2-1.6-1.6-3.9-2.3-5.2-1zm-10.8-8.1c-.7 1.3.3 2.9 2.3 3.9 1.6 1 3.6.7 4.3-.7.7-1.3-.3-2.9-2.3-3.9-2-.6-3.6-.3-4.3.7zm32.4 35.6c-1.6 1.3-1 4.3 1.3 6.2 2.3 2.3 5.2 2.6 6.5 1 1.3-1.3.7-4.3-1.3-6.2-2.2-2.3-5.2-2.6-6.5-1zm-11.4-14.7c-1.6 1-1.6 3.6 0 5.9 1.6 2.3 4.3 3.3 5.6 2.3 1.6-1.3 1.6-3.9 0-6.2-1.4-2.3-4-3.3-5.6-2z"></path></svg> para el control de versiones --- ## Organización del proyecto - La función del **README** es explicar la organización del proyecto y que otros entiendan su contenido -- - Los datos deben de almacenarse en un directorio especial (considerar espacio en disco): -- - Considerar almacenar los datos en un repositorio como respaldo [OSF](https://osf.io/) <svg viewBox="0 0 512 512" style="height:1em;position:relative;display:inline-block;top:.1em;" xmlns="http://www.w3.org/2000/svg"> <g label="icon" id="layer6" groupmode="layer"> <path id="path2" d="m 255.9997,7.9999987 c -34.36057,0 -62.21509,27.8545563 -62.21509,62.2151643 0,20.303056 9.87066,38.160947 24.91769,49.517247 0.18814,-20.457899 16.79601,-36.993393 37.29685,-36.993393 20.50082,0 37.11091,16.535494 37.29909,36.993393 15.04533,-11.3563 24.9177,-29.212506 24.9177,-49.517247 C 318.21272,35.854555 290.35915,7.9999987 255.99915,7.9999987 Z M 293.29654,392.2676 c -0.18814,20.4601 -16.79601,36.99338 -37.29684,36.99338 -20.50082,0 -37.10922,-16.53551 -37.29684,-36.99338 -15.04759,11.35627 -24.91769,29.21246 -24.91769,49.51722 0,34.36059 27.85453,62.21518 62.2151,62.21518 34.36056,0 62.21508,-27.85459 62.21508,-62.21518 0,-20.30306 -9.87066,-38.16095 -24.91767,-49.51722 z M 441.78489,193.78484 c -20.30301,0 -38.16309,9.87068 -49.51717,24.91769 20.45786,0.18819 36.99333,16.79605 36.99333,37.29689 0,20.50085 -16.53547,37.11096 -36.9911,37.29916 11.35634,15.04533 29.21249,24.91769 49.51721,24.91769 C 476.14549,318.21327 504,290.35948 504,255.99942 504,221.6394 476.14549,193.78425 441.78489,193.78425 Z M 82.738898,255.99997 c 0,-20.50139 16.535509,-37.11096 36.993392,-37.29689 -11.35632,-15.04756 -29.214164,-24.91769 -49.517197,-24.91769 -34.36057,0 -62.2150945,27.85455 -62.2150945,62.21517 0,34.3606 27.8545245,62.21516 62.2150945,62.21516 20.303033,0 38.160877,-9.87068 49.517197,-24.91773 -20.457883,-0.18818 -36.993391,-16.796 -36.993391,-37.29688 z M 431.3627,80.636814 c -24.29549,-24.295544 -63.68834,-24.295544 -87.9844,0 -14.35704,14.357057 -20.00298,33.963346 -17.39331,52.633806 -0.0824,0.0809 -0.18198,0.13437 -0.26434,0.21491 -14.578,14.57799 -14.578,38.21689 0,52.79488 14.57797,14.57799 38.21681,14.57799 52.79484,0 0.0824,-0.0824 0.13455,-0.18198 0.21732,-0.26434 18.66819,2.60796 38.27445,-3.03799 52.63151,-17.39336 24.29378,-24.29778 24.29378,-63.68837 -0.003,-87.986153 z M 186.2806,378.51178 c 14.57798,-14.57799 14.57798,-38.21461 0,-52.79319 -14.57798,-14.57853 -38.21683,-14.57798 -52.79481,0 -0.0825,0.0824 -0.13448,0.18199 -0.21476,0.26215 -18.67046,-2.60795 -38.276723,3.03572 -52.63376,17.39505 -24.297753,24.29552 -24.297753,63.6884 0,87.98449 24.29551,24.29552 63.68833,24.29552 87.98439,0 14.35702,-14.35703 20.00297,-33.96333 17.39333,-52.63386 0.0848,-0.0786 0.18364,-0.13228 0.26672,-0.21505 z m 0,-245.02583 c -0.0826,-0.0824 -0.18198,-0.13436 -0.26445,-0.21494 2.60795,-18.66823 -3.038,-38.27452 -17.39332,-52.633811 -24.29777,-24.295544 -63.68832,-24.295544 -87.984405,0 -24.297747,24.297781 -24.297747,63.688381 0,87.986151 14.357042,14.35706 33.963315,20.00301 52.631515,17.39336 0.0808,0.0824 0.13447,0.18199 0.21475,0.26434 14.57799,14.57799 38.21684,14.57799 52.79482,0 14.57797,-14.57802 14.57797,-38.21689 0,-52.79488 z m 245.0821,209.89048 c -14.35703,-14.35703 -33.96329,-20.00301 -52.63378,-17.39505 -0.0809,-0.0824 -0.13228,-0.18199 -0.21506,-0.26215 -14.57797,-14.57799 -38.21685,-14.57799 -52.79482,0 -14.57797,14.57799 -14.57797,38.21461 0,52.79316 0.0827,0.0828 0.18198,0.13455 0.26434,0.21505 -2.60796,18.67053 3.03802,38.27683 17.39334,52.63386 24.29552,24.29552 63.68834,24.29552 87.98439,0 24.29775,-24.29552 24.29775,-63.68841 0.003,-87.98451 z" style="stroke-width:0.07717"></path> </g></svg> -- - **El código es una de las partes más importantes del proyecto** --- ## Organización del proyecto ## Código - Enumerar los scripts de acuerdo al orden en que serán ejecutados .tiny[ ``` +-- 1.QualityControl.sh +-- 2.Trimming.sh +-- 3.ReadAlignment.sh +-- 4.DifferentialExpression.R ``` ] - Utilizar rutas relativas hacia los archivos *input* .tiny[ ``` fastqc ../Data/*.fastqc -o ../Results/ ``` ] - Comentar el script -> facilita que sea entendido por humanos .tiny[ ``` #Change the location of bam files mv ../Data/*.bam ../Results/ ``` ] **Checa el paquete [`here`](https://github.com/jennybc/here_here) desarrollado por Jenny B. - ¡Mejorar el código! -> *Less is more* --- .center[] --- ## Organización del proyecto <img src="./Images/Proyecto.png" width="450px" style="display: block; margin: auto;" /> --- ## Experimento Número de acceso GEO: GSE152699 <img src="./Images/Experiment_ver.png" width="800px" style="display: block; margin: auto;" /> --- class: inverse, center, middle # 3: Flujo de trabajo para el análisis de RNA-seq --- ## Flujo general de trabajo <img src="https://biocorecrg.github.io/RNAseq_course_2019/images/RNAseq_workflow.png" width="550px" style="display: block; margin: auto;" /> .tiny[Imagen tomada de [aquí](https://biocorecrg.github.io/RNAseq_course_2019/salmon.html)] --- ## Primeros pasos <img src="./Images/step1.png" width="540px" style="display: block; margin: auto;" /> --- ## Alineamiento y Quantificación <img src="./Images/step2.png" width="950px" style="display: block; margin: auto;" /> --- ## Expresión Diferencial y análisis funcionales <img src="./Images/step3.png" width="750px" style="display: block; margin: auto;" /> --- ## Flujo general de trabajo <img src="https://biocorecrg.github.io/RNAseq_course_2019/images/RNAseq_workflow.png" width="550px" style="display: block; margin: auto;" /> .tiny[Imagen tomada de [aquí](https://biocorecrg.github.io/RNAseq_course_2019/salmon.html)] --- class: inverse, center, middle # 4: Tipos de archivos --- ## FastQ -- - Derivado del formato FASTA -- - Id secuencia + **Calidad** -- - Generalmente no está comprimido (.fastq) > comprimir (.fastq.gz) -- - Comprende 4 líneas por secuencia: * @ ID del read + información de la corrida * Secuencia * Símbolo "+" * Calidad (Escala **Phred** y código **ASCII**) --- ## FastQ ## Identificador -- - El identificador de la secuencia es la primera línea del archivo .fastq -- - Plataformas de Illumina superiores a 1.8 contienen la siguiente información: -- **@machneID:run number:flowCell ID:lane:tile:read:control number:index** ``` ## gzcat: can't stat: ../Data/s1_R1.fastq.gz (../Data/s1_R1.fastq.gz.gz): No such file or directory ``` --- ## FastQ ## Calidad La calidad de cada base secuenciada, en la escala **Phred**, se calcula de acuerdo la siguiente ecuación: .full-width[.content-box-blue[$$Q = -10 * log10(p)$$]] -- Q = Calidad -- p = Probabilidad de que el *base call* sea incorrecto -- Valores de **p** cercanos a cero -> Alta calidad --- ## FastQ ## Calidad <img src="./Images/Fastqc.png" width="700px" style="display: block; margin: auto;" /> --- ## FastQ ## Calidad -- - Para evitar colocar los valores numéricos de la calidad en el archivo .fastq y saturarlo de información -> **[ASCII](https://www.ascii-code.com/)** -- - Phred + 33 -> Plataformas de Illumina a partir de 1.8 (símbolos especiales + alfanuméricos) -- - Phred + 64 -> Plataformas de Illumina anteriores a 1.8 --- ## FastQ <img src="./Images/ASCII0.png" width="600px" style="display: block; margin: auto;" /> <img src="./Images/ASCII33.png" width="600px" style="display: block; margin: auto;" /> <img src="./Images/ASCII64.png" width="600px" style="display: block; margin: auto;" /> .font50[[ASCII Code - The extended ASCII table](https://www.ascii-code.com/)] --- ## FastQ <img src="./Images/Fastq.png" width="700px" style="display: block; margin: auto;" /> --- class: inverse, center, middle # 5: Control de calidad de los datos --- ## ¿Porqué es importante revisar los datos crudos? -- .full-width[.content-box-blue[Para verificar la calidad de los datos (*QC: quality control*)]] -- - Los datos influencian los análisis posteriores -- .pull-left[ <img src="https://miro.medium.com/max/470/1*DVLpt3zCNaqeZOGNpgME7Q.png" width="150px" style="display: block; margin: auto;" /><img src="https://i.pinimg.com/originals/30/66/ac/3066ac69ae68ac200ace0ca8fe3882c3.jpg" width="150px" style="display: block; margin: auto;" /> ] .pull-right[ <img src="https://memegenerator.net/img/instances/68955295.jpg" width="200px" style="display: block; margin: auto;" /> ] --- ## FastQC Usaremos el programa de [fastQC](https://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc) para checar la calidad de los datos. -- Podemos usar la interfaz gráfica o la línea de comandos, en este caso usaremos la línea de comandos. <img src="https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc.png" width="400px" style="display: block; margin: auto;" /> --- .center2[] --- ### Pasos para installar FastQC: .scroll-output[ 1- Descargar el programa de su página web [fastQC](https://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc) 2- Hacer el archivo ejecutable ```bash # chmod 755 /Applications/FastQC.app/Contents/MacOS/fastqc ls -l /Applications/FastQC.app/Contents/MacOS/fastqc ``` 3- Crear una liga simbolica para ejecutar `fastqc` desde cualquier ubicación ```bash ln -s /Applications/FastQC.app/Contents/MacOS/fastqc /usr/local/bin/fastqc ``` 4- Correr `fastqc` ```bash ##Imprimir el manual del programa usando el flag help fastqc --help ``` ] --- ### Pasos para correr FastQC: .scroll-output[ 1- Checar los datos fastq ```bash pwd ls ../data/Archivos_fastq ``` 2- Crear un directorio para guardar el output ```bash mkdir ../output/QC ``` 3- Correr `FastQC` ```bash # la opción -o es para elegir el directorio de salida fastqc ../data/Archivos_fastq/*.fastq.gz -o ../output/QC/ ``` 4- Checar los archivos generados ```bash #Buscar los archivos generados por fastqc en el directorio de resultados ls ../output/QC ``` ] --- ## Buena calidad El eje x = longitud de la read, y = calidad (phred score) Los datos a continuación muestran reads de buena calidad (Verde = buena, amarilla = aceptable, roja = baja). <img src="https://gwu-omics2019.readthedocs.io/en/latest/_images/fastqc_good.png" width="400px" style="display: block; margin: auto;" /> --- ## Mala calidad ¿Hasta donde los datos son de buena calidad? ¿Estos datos los usarías? <img src="https://gwu-omics2019.readthedocs.io/en/latest/_images/fastqc_bad.png" width="550px" style="display: block; margin: auto;" /> --- ## Ejercicio ¿Cómo se ven nuestros datos? --- ## MultiQC .scroll-output[ [MultiQC](https://multiqc.info/) te permite juntar los output en un solo archivo y poder visualizarlos mejor. *Esta parte es opcional, pero si trabajas con RNA-seq te puede intersar. 1. La instalación se hace con conda, así que primero activamos un ambiente conda ```bash conda activate base pip install multiqc ``` 2. Correr MultiQC ```bash cd ../output/. multiQC . ``` 3. Checar el [output.html](file:///Users/user/Documents/Doctorado/Courses/Taught/minicurso_abr_2021/output/QC/multiqc_report.html#fastqc_per_base_sequence_content) ] --- class: inverse, center, middle # 6: Alineamiento --- <img src="./Images/RNAseq_workflow2.png" width="600px" style="display: block; margin: auto;" /> --- ## Alineamiento <img src="./Images/ReadAlignment.png" width="600px" style="display: block; margin: auto;" /> Buscar la región del genoma/transcriptoma a partir de la cual se originaron los reads --- ## Alineamiento - Indexar genoma/transcriptoma (FASTA, GTF3/GFF3) <img src="./Images/GenomeIndexing.png" width="500px" style="display: block; margin: auto;" /> -- - Alinear los reads (Fastq) al genoma/transcriptoma indexado <img src="./Images/ReadAlignment1.png" width="500px" style="display: block; margin: auto;" /> Imágenes tomadas de [aquí](https://almob.biomedcentral.com/articles/10.1186/s13015-016-0069-5/figures/1) y [aquí](https://figshare.com/articles/figure/_Comparison_of_stranded_and_unstranded_RNA_seq_library_methods_and_their_influence_on_interpretation_and_analysis_/1504417/1) --- ## Alineamiento ## Spliced reads <img src="./Images/SplicedReads2.png" width="600px" style="display: block; margin: auto;" /> .font50[[HISAT: a fast spliced aligner with low memory requirements](https://www.nature.com/articles/nmeth.3317)] --- ## Splice aware - TopHat 2 <img src="./Images/SpliceAware.png" width="600px" style="display: block; margin: auto;" /> -- - HISAT <img src="./Images/HISAT.png" width="600px" style="display: block; margin: auto;" /> -- - STAR <img src="./Images/STAR.png" width="400px" style="display: block; margin: auto;" /> .font50[[Imágenes tomadas de aqui](https://training.galaxyproject.org/training-material/topics/transcriptomics/tutorials/rb-rnaseq/tutorial.html)] --- ## Alineamiento ## Pseudo-alineadores Pseudo-alineadores (quasi-alineadores): - Kallisto - Sailfish - **Salmon** <!-- --> --- ## Alineamiento ## Pseudo-alineadores <img src="./Images/Pseudoalignment.png" width="600px" style="display: block; margin: auto;" /> .font50[[RapMap: a rapid, sensitive and accurate tool for mapping RNA-seq reads to transcriptomes](https://academic.oup.com/bioinformatics/article/32/12/i192/2288985)] --- ## Alineamiento ## Pseudo-alineadores <img src="./Images/PseudoAlignment1.png" width="500px" style="display: block; margin: auto;" /> -- <img src="./Images/PseudoAlignment2.png" width="500px" style="display: block; margin: auto;" /> --- ## Alineamiento ## Pseudo-alineadores <img src="./Images/PseudoAlignment3.png" width="600px" style="display: block; margin: auto;" /> .font50[[Near-optimal probabilistic RNA-seq quantification](https://www.nature.com/articles/nbt.3519?WT.feed_name=subjects_genome-informatics)] --- .center2[] --- ## Generar índice del transcriptoma .scroll-output[ Para ello vamos a requerir: - Archivo **Fasta** del transcriptoma de referencia del humano. Descargado del sitio de [GeneCode](https://www.gencodegenes.org/human/) Lo pueden encontrar en el folder de `Transcriptome/` - Código para genear el índice ``` salmon index -t --genecode path_to_transcriptome.fa.gz -i path_to_save_index ``` `-t`: Ubicación al archivo Fasta del transcriptoma `-i`: Ubicación para salvar el índice `--genecode`: El Fasta del transcriptoma de referencia está en formato de GENECODE ] --- ## Cuantificar la abundancia de los transcritos .scroll-output[ Requerimientos: - Archivos **Fastq** de las lecturas -> Paired end R1 y R2 Ubicados en el folder de `data` - Índice generado en el paso anterior - Código para producir cuantificar los transcritos ``` salmon quant -i path_to_index -l A -1 path_to_R1 -2 path_to_R2 -o path_to_store_results ``` `-i`: Ubicación del índice `-l`: Tipo de librería `-1 y -2`: Ubicación a las lecturas R1 y R2 `-o`: Ubicación para almacenar los resultados ```bash ##Crear un directorio para almacenar los datos de los conteos mkdir -p ../salmon_quant ##Llamar salmon para realizar el conteo salmon quant -i ../transcriptome/genecode.v37.salmon141 \ -l A \ -1 ../Data/s1_R1.fastq.gz -2 ../Data/s1_R2.fastq.gz \ -p 6 --validateMappings \ -o ../salmon_quant/s1_quant ``` ] --- class: inverse, center, middle # 7: Tximeta y Tximport --- ## Recordatorio ¿Dónde estamos? <img src="https://biocorecrg.github.io/RNAseq_course_2019/images/RNAseq_workflow.png" width="450px" style="display: block; margin: auto;" /> .tiny[Imagen tomada de [aquí](https://biocorecrg.github.io/RNAseq_course_2019/salmon.html)] --- ## Recordatorio ¿Dónde estamos? <img src="Images/worflowTximeta.png" width="450px" style="display: block; margin: auto;" /> .tiny[Imagen tomada de [aquí](https://biocorecrg.github.io/RNAseq_course_2019/salmon.html)] --- ## Tximeta Paquetería que conserva <img src="https://journals.plos.org/ploscompbiol/article/figure/image?size=large&id=10.1371/journal.pcbi.1007664.g001" width="650px" style="display: block; margin: auto;" /> .tiny[[Love, Michael I., et al. "Tximeta: Reference sequence checksums for provenance identification in RNA-seq." PLoS computational biology 16.2 (2020): e1007664.](https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007664)] --- ## Importar datos Utilizamos `tximeta` para importar los datos. .content-box-red[ Nota: `tximeta` espera dos columnas: - `files`: con la rut a `quant.sf` - `names`: con los nombres de las muestras ] --- .center2[] --- ## Importar datos .scroll-output[ Importamos la información de cada muestra ```r ##Leemos los datos coldata <- read.delim(here::here("output/salmon_quants/metadata.txt")) ##Generamos la columna "names" coldata <- coldata %>% dplyr::rename(names = unique_id) coldata ``` ``` ## Sample Cell Treatment Replicate names ## 1 s1 A Control 1 A_Control_1 ## 2 s2 A Control 2 A_Control_2 ## 3 s3 A Control 3 A_Control_3 ## 4 s4 M Control 1 M_Control_1 ## 5 s5 M Control 2 M_Control_2 ## 6 s6 M Control 3 M_Control_3 ## 7 s7 A Verafinib 1 A_Verafinib_1 ## 8 s8 A Verafinib 2 A_Verafinib_2 ## 9 s9 A Verafinib 3 A_Verafinib_3 ## 10 s10 M Verafinib 1 M_Verafinib_1 ## 11 s11 M Verafinib 2 M_Verafinib_2 ## 12 s12 M Verafinib 3 M_Verafinib_3 ``` ```r ##Ahora las cuentas generadas por `Salmon`, para ello ##Ubicamos el dir de trabajo dir <- file.path(here::here("output/salmon_quants")) ##Verificar que estén los directorios list.files(dir) ``` ``` ## [1] "Icon\r" "metadata.txt" "s1_quant" "s10_quant" "s11_quant" ## [6] "s12_quant" "s2_quant" "s3_quant" "s4_quant" "s5_quant" ## [11] "s6_quant" "s7_quant" "s8_quant" "s9_quant" ``` ```r ##¿¿Que hacemos aqui?? Verificar que el path se generó correctamente #file.path(dir,paste0(info$Sample,"_quant"),"quant.sf") coldata$files <- file.path(dir,paste0(coldata$Sample,"_quant"),"quant.sf") ##Corroboramos que los archivos existan data.frame(coldata$Sample, file.exists(coldata$files)) ``` ``` ## coldata.Sample file.exists.coldata.files. ## 1 s1 TRUE ## 2 s2 TRUE ## 3 s3 TRUE ## 4 s4 TRUE ## 5 s5 TRUE ## 6 s6 TRUE ## 7 s7 TRUE ## 8 s8 TRUE ## 9 s9 TRUE ## 10 s10 TRUE ## 11 s11 TRUE ## 12 s12 TRUE ``` Importar los datos usando `tximeta` ```r #BiocManager::install("tximeta") library("tximeta") se <- tximeta(coldata) ``` ] --- class: inverse, center, middle # 8: Objeto Summarized Experiment --- .center2[ <img src="./Images/Slots.png" width="700px" /> ] --- ## Summarized Experiment <img src="./Images/Summarized_experiment.png" width="400px" style="display: block; margin: auto;" /> --- ## Summarized Experiment <img src="./Images/ColData.png" width="400px" style="display: block; margin: auto;" /> --- ## Summarized Experiment ## ColData .scroll-output[ - El cajón o slot correspondiente a `ColData` contiene la tabla de metadatos `coldata` creada para importar las cuentas con tximeta - Para acceder a **.red[ColData]** usar el siguiente comando: ```r colData(se) ``` ``` ## DataFrame with 12 rows and 6 columns ## Sample Cell Treatment Replicate names ## <character> <character> <character> <integer> <character> ## A_Control_1 s1 A Control 1 A_Control_1 ## A_Control_2 s2 A Control 2 A_Control_2 ## A_Control_3 s3 A Control 3 A_Control_3 ## M_Control_1 s4 M Control 1 M_Control_1 ## M_Control_2 s5 M Control 2 M_Control_2 ## ... ... ... ... ... ... ## A_Verafinib_2 s8 A Verafinib 2 A_Verafinib_2 ## A_Verafinib_3 s9 A Verafinib 3 A_Verafinib_3 ## M_Verafinib_1 s10 M Verafinib 1 M_Verafinib_1 ## M_Verafinib_2 s11 M Verafinib 2 M_Verafinib_2 ## M_Verafinib_3 s12 M Verafinib 3 M_Verafinib_3 ## Condition ## <factor> ## A_Control_1 Control ## A_Control_2 Control ## A_Control_3 Control ## M_Control_1 Control ## M_Control_2 Control ## ... ... ## A_Verafinib_2 Verafinib ## A_Verafinib_3 Verafinib ## M_Verafinib_1 Verafinib ## M_Verafinib_2 Verafinib ## M_Verafinib_3 Verafinib ``` ] --- ## Summarized Experiment ## ColData - El slot **ColData** es un objeto con clase de *DataFrame* .code70[ ```r class(colData(se)) ``` ``` ## [1] "DFrame" ## attr(,"package") ## [1] "S4Vectors" ``` ] -- - *Rownames* de **ColData** corresponden a los *Colnames* en el slot **Assay**- **.red[Importante para análisis con DESeq2]** .code70[ ```r rownames(colData(se)) ``` ``` ## [1] "A_Control_1" "A_Control_2" "A_Control_3" "M_Control_1" ## [5] "M_Control_2" "M_Control_3" "A_Verafinib_1" "A_Verafinib_2" ## [9] "A_Verafinib_3" "M_Verafinib_1" "M_Verafinib_2" "M_Verafinib_3" ``` ] --- ## Summarized experiment <img src="./Images/RowRanges.png" width="400px" style="display: block; margin: auto;" /> --- ## Summarized experiment ## rowRanges .scroll-output[ - El cajón de `rowRanges` hace referencia a las coordenadas de cada transcrito - Para acceder al **.red[rowRanges]** usar: ```r rowRanges(se) ``` ``` ## GRanges object with 172103 ranges and 6 metadata columns: ## seqnames ranges strand | tx_id ## <Rle> <IRanges> <Rle> | <character> ## ENST00000415118 14 22438547-22438554 + | ENST00000415118 ## ENST00000434970 14 22439007-22439015 + | ENST00000434970 ## ENST00000448914 14 22449113-22449125 + | ENST00000448914 ## ENST00000604642 15 20003840-20003862 - | ENST00000604642 ## ENST00000603326 15 20004797-20004815 - | ENST00000603326 ## ... ... ... ... . ... ## ENST00000444456 1 247974741-247975653 + | ENST00000444456 ## ENST00000416377 1 248003129-248004068 + | ENST00000416377 ## ENST00000318104 1 248121932-248122882 + | ENST00000318104 ## ENST00000606902 1 248498308-248498649 + | ENST00000606902 ## ENST00000427827 1 248549444-248549762 - | ENST00000427827 ## tx_biotype tx_cds_seq_start tx_cds_seq_end ## <character> <integer> <integer> ## ENST00000415118 TR_D_gene 22438547 22438554 ## ENST00000434970 TR_D_gene 22439007 22439015 ## ENST00000448914 TR_D_gene 22449113 22449125 ## ENST00000604642 IG_D_gene 20003840 20003862 ## ENST00000603326 IG_D_gene 20004797 20004815 ## ... ... ... ... ## ENST00000444456 unprocessed_pseudogene <NA> <NA> ## ENST00000416377 unprocessed_pseudogene <NA> <NA> ## ENST00000318104 unprocessed_pseudogene <NA> <NA> ## ENST00000606902 unprocessed_pseudogene <NA> <NA> ## ENST00000427827 unprocessed_pseudogene <NA> <NA> ## gene_id tx_name ## <character> <character> ## ENST00000415118 ENSG00000223997 ENST00000415118 ## ENST00000434970 ENSG00000237235 ENST00000434970 ## ENST00000448914 ENSG00000228985 ENST00000448914 ## ENST00000604642 ENSG00000270961 ENST00000604642 ## ENST00000603326 ENSG00000271317 ENST00000603326 ## ... ... ... ## ENST00000444456 ENSG00000237492 ENST00000444456 ## ENST00000416377 ENSG00000232215 ENST00000416377 ## ENST00000318104 ENSG00000177233 ENST00000318104 ## ENST00000606902 ENSG00000271934 ENST00000606902 ## ENST00000427827 ENSG00000227102 ENST00000427827 ## ------- ## seqinfo: 47 sequences from GRCh38 genome ``` ] --- ## Summarized experiment <img src="./Images/Assay.png" width="400px" style="display: block; margin: auto;" /> --- ## Summarized experiment ## Assay .scroll-output[ - El slot `assay` almacena la información de las cuentas para cada transcrito dividida en tres niveles: ```r assayNames(se) ``` ``` ## [1] "counts" "abundance" "length" ``` - Para acceder a la matriz de cuentas estimadas por *Salmon*, correr: ```r head(assay(se), 5) ``` ``` ## A_Control_1 A_Control_2 A_Control_3 M_Control_1 M_Control_2 ## ENST00000415118 0 0 0 0 0 ## ENST00000434970 0 0 0 0 0 ## ENST00000448914 0 0 0 0 0 ## ENST00000604642 0 0 0 0 0 ## ENST00000603326 0 0 0 0 0 ## M_Control_3 A_Verafinib_1 A_Verafinib_2 A_Verafinib_3 ## ENST00000415118 0 0 0 0 ## ENST00000434970 0 0 0 0 ## ENST00000448914 0 0 0 0 ## ENST00000604642 0 0 0 0 ## ENST00000603326 0 0 0 0 ## M_Verafinib_1 M_Verafinib_2 M_Verafinib_3 ## ENST00000415118 0 0 0 ## ENST00000434970 0 0 0 ## ENST00000448914 0 0 0 ## ENST00000604642 0 0 0 ## ENST00000603326 0 0 0 ``` ] --- ## Summarized experiment ## Assay - Las matriz de abundancia *(TPM)* puede obtenerse: .code70[ ```r ## Obtener matriz de TPM head(se@assays@data$abundance, 5) ``` ``` ## A_Control_1 A_Control_2 A_Control_3 M_Control_1 M_Control_2 ## ENST00000415118 0 0 0 0 0 ## ENST00000434970 0 0 0 0 0 ## ENST00000448914 0 0 0 0 0 ## ENST00000604642 0 0 0 0 0 ## ENST00000603326 0 0 0 0 0 ## M_Control_3 A_Verafinib_1 A_Verafinib_2 A_Verafinib_3 ## ENST00000415118 0 0 0 0 ## ENST00000434970 0 0 0 0 ## ENST00000448914 0 0 0 0 ## ENST00000604642 0 0 0 0 ## ENST00000603326 0 0 0 0 ## M_Verafinib_1 M_Verafinib_2 M_Verafinib_3 ## ENST00000415118 0 0 0 ## ENST00000434970 0 0 0 ## ENST00000448914 0 0 0 ## ENST00000604642 0 0 0 ## ENST00000603326 0 0 0 ``` ] --- ## Summarized experiment .scroll-output[ ¿Recuerdan a qué tiene que ser igual *Rownames* del slot colData? .code90[ ```r rownames(colData(se)) ``` ``` ## [1] "A_Control_1" "A_Control_2" "A_Control_3" "M_Control_1" ## [5] "M_Control_2" "M_Control_3" "A_Verafinib_1" "A_Verafinib_2" ## [9] "A_Verafinib_3" "M_Verafinib_1" "M_Verafinib_2" "M_Verafinib_3" ``` ```r colnames(assay(se)) ``` ``` ## [1] "A_Control_1" "A_Control_2" "A_Control_3" "M_Control_1" ## [5] "M_Control_2" "M_Control_3" "A_Verafinib_1" "A_Verafinib_2" ## [9] "A_Verafinib_3" "M_Verafinib_1" "M_Verafinib_2" "M_Verafinib_3" ``` ```r ## Comprobar que rownames de colData es igual a colnames de assay row.names(colData(se)) == colnames(assay(se)) ``` ``` ## [1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE ``` ] ] --- class: inverse, center, middle # 9: Normalización de los datos --- ## Normalización Es el primer paso del análisis de expresión diferencial y es necesario para hacer comparaciones acertadas entre muestras. -- Las cuentas crudas están conformadas por un componente "interesante" (la expresión de RNA) y componentes "no interesantes" (como los batch effects, ruido de la plataforma, etc.). -- La normalzación escala las cuentas para tratar de reducir los componentes "no interesantes" y poder comparar las muestras entre si. --- ## Criteríos para normalizar Se puede normalizar considerando: - La profundidad (tamaño de librería) <img src="https://hbctraining.github.io/DGE_workshop/img/normalization_methods_depth.png" width="450px" style="display: block; margin: auto;" /> --- ## Criteríos para normalizar Se puede normalizar considerando: - El tamaño del gen <img src="https://hbctraining.github.io/DGE_workshop/img/normalization_methods_length.png" width="450px" style="display: block; margin: auto;" /> --- ## Criteríos para normalizar Se puede normalizar considerando: - Composición de RNA <img src="https://hbctraining.github.io/DGE_workshop/img/normalization_methods_composition.png" width="350px" style="display: block; margin: auto;" /> .tiny[Imagen tomada de [aquí](https://biocorecrg.github.io/RNAseq_course_2019/salmon.html)] --- ## Métodos comunes .scroll-output[ | Método de normalización | Descripción | Factores de evaluación | Recomendaciones de uso | |:-----------------------------------------------------------------------------------:|:----------------------------------------------------------------------------------------------------------------------------:|:--------------------------------------------------:|:-------------------------------------------------------------------------------------------------------------------:| | CPM (cuentas por millón): cuentas escalados por el número total de lecturas | profundidad de secuenciación | comparaciones de cuentas de genes entre réplicas del mismo grupo de muestras| NO para comparaciones dentro de la muestra o análisis de DE.| | TPM (transcritos por kilobase por millón de lecturas): cuentas por longitud de transcripción (kb) por millón de lecturas mapeadas | profundidad de secuenciación y longitud de genes| comparaciones de cuentas de genes dentro de una muestra o entre muestras del mismo grupo de muestras| NO para análisis de DE. | | RPKM/FPKM (lecturas/fragmentos por kilobase de exón por millón de lecturas/fragmentos mapeados| similar a TPM, profundidad de secuenciación y longitud del gen | comparaciones de cuentas entre genes dentro de una muestra | NO para comparaciones entre muestras o análisis de DE.| | Mediana de ratios de DESeq2 | cuentas divididas por factores de tamaño específicos de la muestra determinados por la mediana del ratio de cuentas de genes en relación con la media geométrica por gen | profundidad de secuenciación y composición del RNA | comparaciones de cuentas de genes entre muestras y para el análisis de DE; NO para comparaciones dentro de la muestra | | La media cortada de los valores M de EdgeR (TMM) | utiliza una media recortada ponderada de los ratios de expresión logarítmica entre las muestras | profundidad de secuenciación | composición de RNA y longitud de los genes. | .tiny[ [Tabla tomada de Intro to DGE](https://hbctraining.github.io/DGE_workshop/lessons/02_DGE_count_normalization.html#:~:text=Normalization%20is%20the%20process%20of,between%20and%2For%20within%20samples.) ] ] --- ## DESeq2 - Normalización para análisis de expresión diferencial: - Factor de normalización de `DESeq2` - Normalización para visualización u otras aplicaciones: - Variance stabilizing transformation (VST) - Regularized-logarithm transformation (rlog) --- ## DESeq2 .scroll-output[ `DESeq2`ajusta a un modelo lineal generalizado (GLM) de la familia binomial negativa (NB). ``` ## # A tibble: 2 x 3 ## gene muestraA muestraB ## <chr> <dbl> <dbl> ## 1 gene1 1749 943 ## 2 gene2 35 29 ``` 1. Crea una pseudo-referencia por muestra (promedio geometrico por fila) `sqrt(muestraA * muestra B)` ``` ## # A tibble: 2 x 4 ## # Rowwise: ## gene muestraA muestraB prom_geom ## <chr> <dbl> <dbl> <dbl> ## 1 gene1 1749 943 1284. ## 2 gene2 35 29 31.9 ``` 2. Se calcula la fración `muestra/pseudo-referencia` ``` ## # A tibble: 2 x 6 ## # Rowwise: ## gene muestraA muestraB prom_geom muestraA_pseudo_ref muestraB_pseudo_ref ## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> ## 1 gene1 1749 943 1284. 1.36 0.734 ## 2 gene2 35 29 31.9 1.10 0.910 ``` 3. Se calcula un factor de normalización (size factor) utilizando la `mediana` por columnas. ``` ## [1] 1.230235 ``` ``` ## [1] 0.8222689 ``` 4. Se dividen las `cuentas crudas/size factor` para calcular las cuentas normalizadas. ``` ## # A tibble: 2 x 3 ## # Rowwise: ## gene muestraA muestraB ## <chr> <dbl> <dbl> ## 1 gene1 1749 943 ## 2 gene2 35 29 ``` ``` ## # A tibble: 2 x 2 ## # Rowwise: ## norm_muestraA norm_muestraB ## <dbl> <dbl> ## 1 1422. 1147. ## 2 28.4 35.3 ``` ] --- .center2[] --- ## Ejercicio .scroll-output[ Se pueden ocupar otras transformaciones en la paquetería de `DESeq2` pero no son las mas recomendadas para DE. Para normalizar, primero construimos el objeto `DESeqDataSet`, para ello necesitamos definir el modelo de DE (Quienes son Controles y quienes tratamiento). ```r # Importamos las librerías que necesitemos library("DESeq2") library("dplyr") library("ggplot2") library("vsn") ``` ```r # Definir los grupos que se desean comparar se$Condition <- factor(se$Condition) # Checar cantidad de muestras table(se$Condition) # Generar el objeto de DESeq dds <- DESeqDataSet(se, design = ~ Condition) # Prefiltrado para quitar genes con cuentas bajas keep <- rowSums(counts(dds)) >= 6 dds <- dds[keep, ] # Funcion que normaliza los datos y realiza el analisis de expresión differencial dds <- DESeq(dds) ``` ] --- ## Otras transformaciones .scroll-output[ Puedes realizar otras transformaciones en `DESeq2` para estabilizar la varianza a través de los differentes valores promedio de expresión. 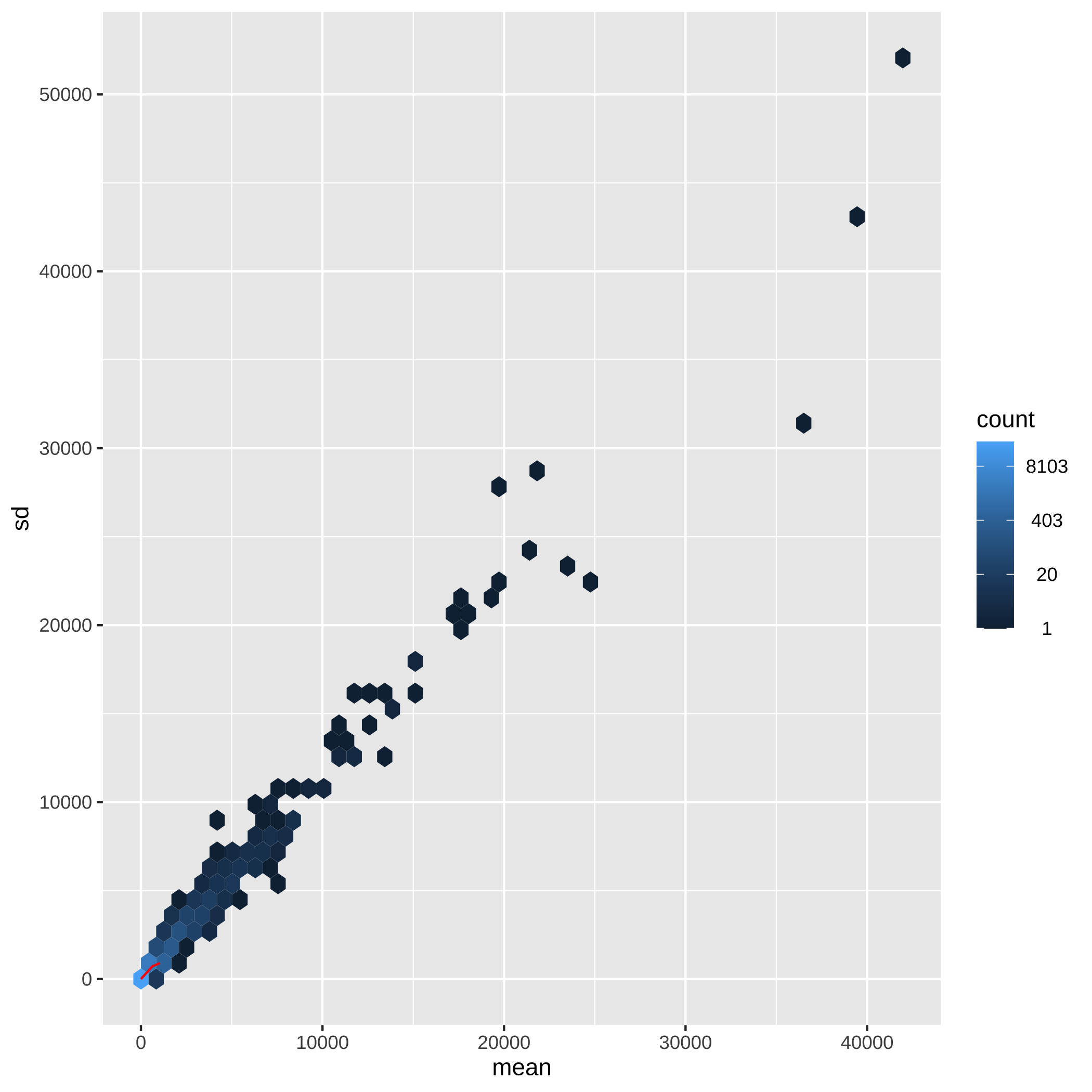<!-- -->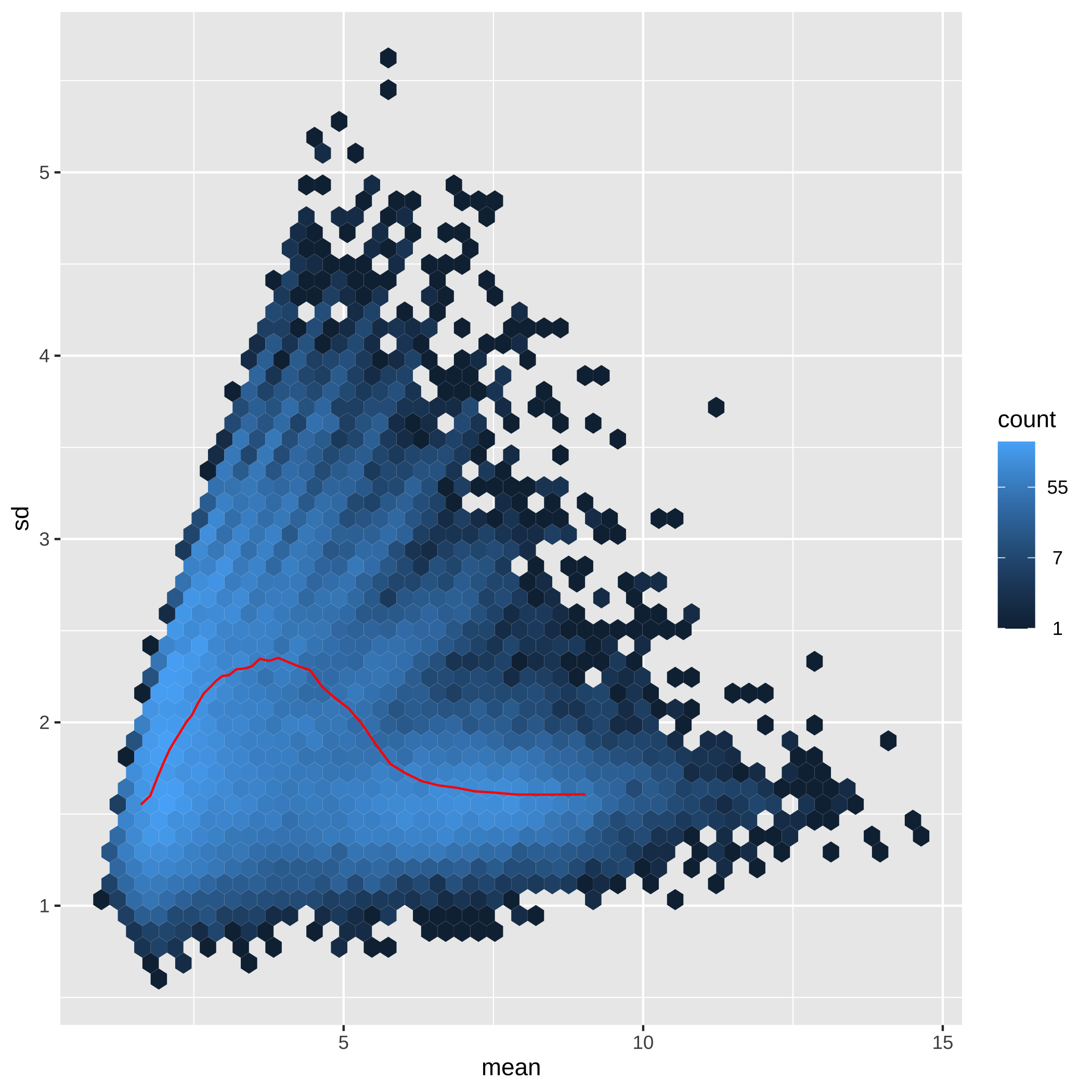<!-- --> ```r # variance stabilizing transformation (VST), (Anders and Huber 2010) vsd <- vst(dds, blind = FALSE) head(assay(vsd), 3) ``` ``` ## A_Control_1 A_Control_2 A_Control_3 M_Control_1 M_Control_2 ## ENST00000641506 5.243738 5.022432 5.236241 5.121340 5.055195 ## ENST00000611116 5.058149 5.063223 4.758866 4.737813 4.871766 ## ENST00000390289 4.900795 5.154803 5.002771 4.909267 4.423150 ## M_Control_3 A_Verafinib_1 A_Verafinib_2 A_Verafinib_3 ## ENST00000641506 5.176386 5.508017 5.293662 5.474252 ## ENST00000611116 5.289621 4.874653 4.423150 4.423150 ## ENST00000390289 5.040044 5.058810 4.423150 5.039269 ## M_Verafinib_1 M_Verafinib_2 M_Verafinib_3 ## ENST00000641506 5.716058 5.895262 5.482647 ## ENST00000611116 5.185938 5.045729 5.180529 ## ENST00000390289 5.571450 5.182835 5.294481 ``` ```r # regularized-logarithm transformation (rlog), (Love, Huber, and Anders 2014) rld <- rlog(dds, blind = FALSE) head(assay(rld), 3) ``` ``` ## A_Control_1 A_Control_2 A_Control_3 M_Control_1 M_Control_2 ## ENST00000641506 2.905283 2.239445 2.8848975 2.5548691 2.4040501 ## ENST00000611116 1.829812 1.846142 0.6284644 0.5186003 1.1642110 ## ENST00000390289 1.416904 2.289820 1.8015912 1.4496085 0.1791245 ## M_Control_3 A_Verafinib_1 A_Verafinib_2 A_Verafinib_3 ## ENST00000641506 2.739348 3.526028 3.03947595 3.45477910 ## ENST00000611116 2.444677 1.174766 -0.05979834 -0.07010645 ## ENST00000390289 1.918379 1.975739 0.15929486 1.91598057 ## M_Verafinib_1 M_Verafinib_2 M_Verafinib_3 ## ENST00000641506 3.938412 4.256634 3.472666 ## ENST00000611116 2.166800 1.752071 2.152501 ## ENST00000390289 3.267274 2.339360 2.635467 ``` ```r # Normalización con el factor de normalizacion dds <- estimateSizeFactors(dds) ``` ``` ## using 'avgTxLength' from assays(dds), correcting for library size ``` ```r # Juntar los datos de las tres normalizaciones df <- bind_rows( as_data_frame(log2(counts(dds, normalized=TRUE)[, 1:2]+1)) %>% mutate(transformation = "log2(x + 1)"), as_data_frame(assay(vsd)[, 1:2]) %>% mutate(transformation = "vst"), as_data_frame(assay(rld)[, 1:2]) %>% mutate(transformation = "rlog")) ``` ``` ## Warning: `as_data_frame()` was deprecated in tibble 2.0.0. ## Please use `as_tibble()` instead. ## The signature and semantics have changed, see `?as_tibble`. ``` ```r # Renombrar columnas colnames(df)[1:2] <- c("x", "y") # Nombre de las graficas lvls <- c("log2(x + 1)", "vst", "rlog") # Agrupar los tres tipos de normalizacion en grupos como factores df$transformation <- factor(df$transformation, levels=lvls) # Plotear los datos ggplot(df, aes(x = x, y = y)) + geom_hex(bins = 80) + coord_fixed() + facet_grid( . ~ transformation) ``` 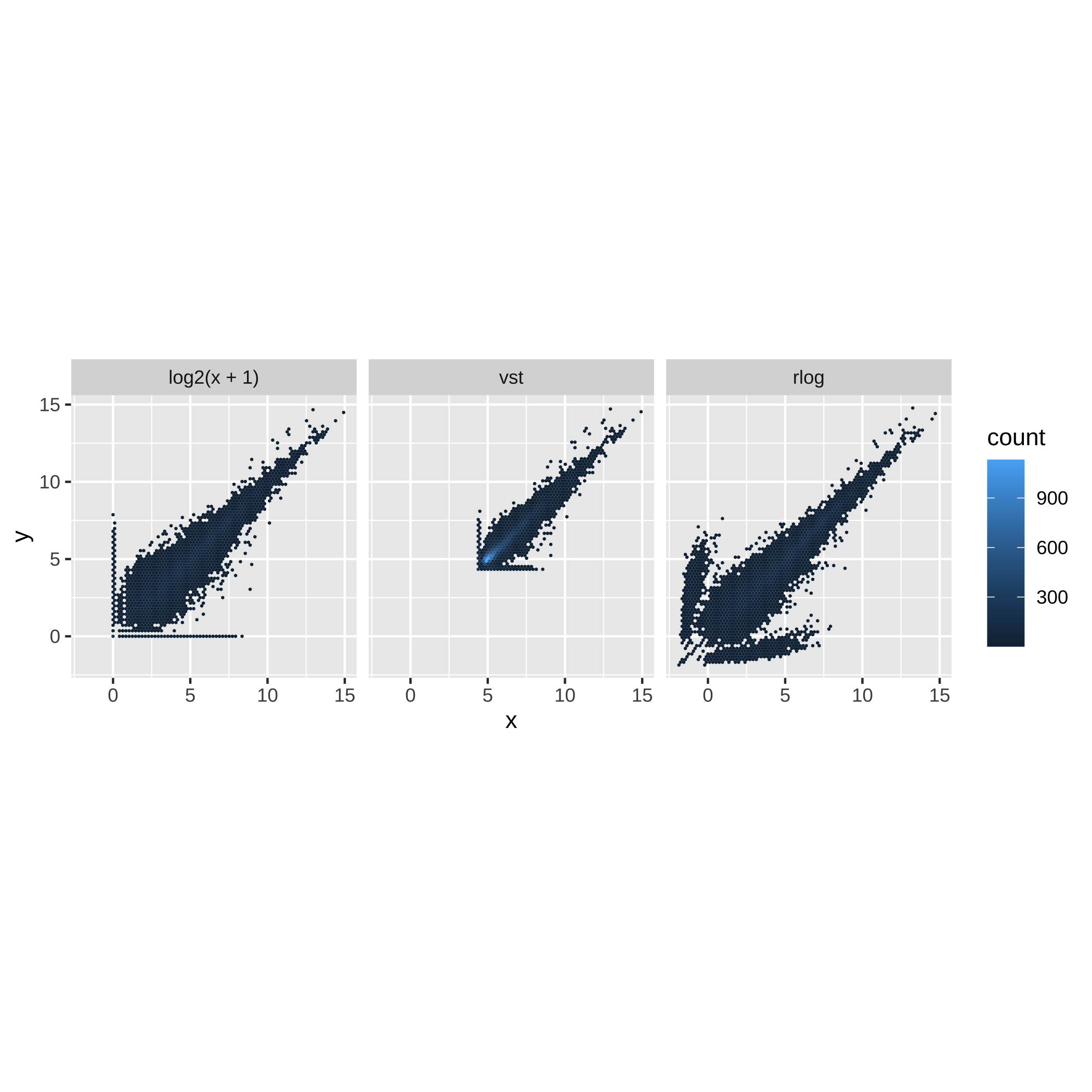<!-- --> ] --- class: inverse, center, middle # 10: Exploración de los datos --- .center2[ <img src="./Images/PCAplot.png" width="700px" /> ] --- ## Exploración de datos ## ¿Por qué es importante explorar los datos? -- - Paso previo al análisis de expresión diferencial -- - Análisis de calidad de los datos -- - Permite conocer la congurencian entre individuos o réplicas -- - Mediante gráficos, visualizar comportamiento de los datos -> Outliers --- ## Exploración de datos .pull-left[ <img src="./Images/PCAplot2.png" width="400px" /> ] -- .pull-right[ <img src="./Images/Heatmap.png" width="400px" /> ] --- ## Exploración de datos .pull-left[ <img src="./Images/PCAplot2.png" width="400px" /> **.red[PCA]** ] .pull-right[ <img src="./Images/Heatmap.png" width="400px" /> **.red[Heatmap]** ] --- ## PCA -- - Análisis de componentes principales -- - Método algebraico para reducir la dimensionalidad de sets de datos complejos (múltiples variables) -- - Reducción de dimensionalidad o variables permite un análisis exploratorio más intuitivo -- .content-box-red[Reducción de variables implica preservar y captar la mayor información sobre los datos ] --- ## PCA ## Normalización de los datos -- <img src="./Images/zscore.png" width="350px" style="display: block; margin: auto;" /> Escalación de los datos -> Normalización para hacerlos más comparables -- <img src="./Images/covariancematrix.png" width="350px" style="display: block; margin: auto;" /> Busqueda de correlación entre las variables -> correlación positiva o negativa --- ## PCA .center[  ] -- .content-box-red[Los componentes principales son los nuevos ejes que maximizan la distancia de los datos al origen ] --- ## PCA ## Componentes principales -- - El .green[primer componente] es aquel en el que los datos presentan la mayor separación (variación) -- - El .orange[segundo componente] es el que tiene la segunda mayor separación entre los datos y es perpendicular al primer componente -- - ¿Cuantos componentes existen? -> tantas variables en el set de datos --- ## PCA ## Componentes principales - Cada componente principal tiene asociado un eigenvector (vector unitario) y un eigenvalue (cantidad escalar) -- - Los eigenvalues son la suma del cuadrado de las distancias de los puntos proyectados sobre dicho componente principal -- - Los eigenvalues permiten seleccionar cuál es el componente principal que explica la mayor variación en los datos --- ## PCA <img src="./Images/Screeplot.png" width="450px" style="display: block; margin: auto;" /> Screeplot --- ## PCA - En sets de datos de RNAseq, el PCA permite visualizar la distancia o congruencia de los datos -- - Generalmente son gráficas de puntos (muestras) en dos dimensiones (dos componentes) que resumen las principales fuentes de varianza -- .center[ <img src="./Images/PCARNAseq.png" width="400px" /> ] --- ## ¿Cómo crear nuestro propio PCA? .scroll-output[ Primero instalen las siguientes librerías: ```r library(DESeq2) library(PCAtools) ``` Usemos la siguiente normalización logarítmica de las cuentas (toma en cuenta el tamaño de la librería) ```r ## Transformación recomendada para sets de con menos de 30 muestras rld <- rlog(dds, blind = F) ``` Si tuvieramos un número mayor de muestras (n > 30) entonces conviene: ```r vsd <- vst(dds, blind = T) ``` ] --- ## PCA .scroll-output[ - Usemos la función interna de *DESeq2* para graficar nuestro PCA: ```r ## El argumento intgroup permite especificar mediante cuál variable colorear los datos plotPCA(rld, intgroup = "Treatment") ``` **DESeq2 evalúa los 500 genes con mayor varianza del set de datos** - Con la librería de *PCAtools*: ```r ## Crear un objeto que contenga los datos del PCA PCA <- pca(assay(rld), scale = T, metadata = colData(rld)) ## Graficar en 2D los resultados biplot(PCA, colby = "Treatment") ## Generar un screeplot para visualizar la varianza asociada a cada componente screeplot(PCA) ``` ] --- .center2[ ] --- ## PCA ## ¿Cómo ven los resultados? -- .pull-left[**DESeq2** <img src="Slides_files/figure-html/unnamed-chunk-100-1.png" width="350px" style="display: block; margin: auto;" /> ] -- .pull-right[**PCA tools** <img src="Slides_files/figure-html/unnamed-chunk-101-1.png" width="350px" style="display: block; margin: auto;" /> ] --- ## PCA ## ¿Cuánta varianza es explicada por cada componente? <img src="Slides_files/figure-html/unnamed-chunk-102-1.png" width="400px" style="display: block; margin: auto;" /> --- ## PCA y MDS .scroll-output[ Generemos un MDS plot interactivo con los datos. Para ello usemos la librería de *Glimma* y la función de `GlimmaMDS` ```r glimmaMDS(dds) ``` ] --- class: inverse, center, middle # 13: Análisis de expresión diferencial --- ## Análisis de expresión diferencial (DE) Paso para obtener cuáles son los genes que varían entre las condiciones <img src="https://raw.githubusercontent.com/hbctraining/DGE_workshop/master/img/de_theory.png" width="750px" style="display: block; margin: auto;" /> --- ## Pasos en el DE <img src="https://github.com/hbctraining/DGE_workshop/raw/master/img/deseq2_workflow_separate.png" width="350px" style="display: block; margin: auto;" /> --- ## Dispersión La dispersión es una medida de la variabilidad de los datos como (varianza, sd, etc.). En `DESeq2` se utiliza `α`: $$ \alpha \propto 1/mean $$ $$ \alpha \propto variance $$ Así que **La dispersión es mayor para cuentas más bajas y menor para cuenas más altas**. Y la **dispersión refleja la varianza**. .full-width[.content-box-red[ `DESeq2` asume que los genes con similar expresión tienen similar dispersión ]] --- ## Ajustando la dispersión <img src="https://github.com/hbctraining/DGE_workshop/raw/master/img/deseq_dispersion1.png" width="450px" style="display: block; margin: auto;" /> --- ## Reduciendo la dispersión <img src="https://github.com/hbctraining/DGE_workshop/raw/master/img/deseq_dispersion2.png" width="750px" style="display: block; margin: auto;" /> --- .center2[] --- ## Ejercicio .scroll-output[ La librería de `DESeq2` normaliza y realiza el análisis de expresión diferencial en una sola función. Así que ya realizamos el análisis, vamos a verlo: ```r library("DESeq2") library("dplyr") library("ggplot2") ``` Para ver los resultados podemos usar la función `results` ```r # Obtener los resultados del DE res <- results(dds) res ``` ``` ## log2 fold change (MLE): Condition Verafinib vs Control ## Wald test p-value: Condition Verafinib vs Control ## DataFrame with 35483 rows and 6 columns ## baseMean log2FoldChange lfcSE stat pvalue ## <numeric> <numeric> <numeric> <numeric> <numeric> ## ENST00000641506 10.05792 1.3610503 0.360096 3.7796900 1.57024e-04 ## ENST00000611116 3.26143 -0.0421221 0.808389 -0.0521062 9.58444e-01 ## ENST00000390289 4.55373 0.9128357 0.601744 1.5169831 1.29271e-01 ## ENST00000361390 3379.18027 2.1135181 0.379169 5.5740792 2.48843e-08 ## ENST00000361453 6522.68449 1.5906491 0.385459 4.1266327 3.68114e-05 ## ... ... ... ... ... ... ## ENST00000622095 7.43638 0.769911 0.620186 1.241418 2.14451e-01 ## ENST00000605551 40.04273 -0.846855 0.397376 -2.131115 3.30797e-02 ## ENST00000579209 9.61591 1.597767 0.386827 4.130447 3.62059e-05 ## ENST00000500626 3.46026 0.798818 0.733797 1.088610 2.76326e-01 ## ENST00000314835 17.48904 0.166453 0.674825 0.246661 8.05171e-01 ## padj ## <numeric> ## ENST00000641506 1.62737e-03 ## ENST00000611116 9.82689e-01 ## ENST00000390289 3.24642e-01 ## ENST00000361390 7.31775e-07 ## ENST00000361453 4.68935e-04 ## ... ... ## ENST00000622095 0.451633725 ## ENST00000605551 0.124474321 ## ENST00000579209 0.000463051 ## ENST00000500626 0.525737614 ## ENST00000314835 0.915847174 ``` ```r summary(res) ``` ``` ## ## out of 35483 with nonzero total read count ## adjusted p-value < 0.1 ## LFC > 0 (up) : 4420, 12% ## LFC < 0 (down) : 4315, 12% ## outliers [1] : 18, 0.051% ## low counts [2] : 0, 0% ## (mean count < 1) ## [1] see 'cooksCutoff' argument of ?results ## [2] see 'independentFiltering' argument of ?results ``` Para ver que hay en cada columna: ```r mcols(res, use.names = TRUE) ``` ``` ## DataFrame with 6 rows and 2 columns ## type description ## <character> <character> ## baseMean intermediate mean of normalized c.. ## log2FoldChange results log2 fold change (ML.. ## lfcSE results standard error: Cond.. ## stat results Wald statistic: Cond.. ## pvalue results Wald test p-value: C.. ## padj results BH adjusted p-values ``` Podemos hacer cortes: ```r resLFC1 <- results(dds, lfcThreshold=1) table(resLFC1$padj < 0.1) ``` ``` ## ## FALSE TRUE ## 34309 1156 ``` ] --- class: inverse, center, middle # 14: Visualización de los resultados --- .center[ ### ¿Cuántos tipos de gráficos conoces para visualizar resultados del análisis DE? ] .pull-left[ <img src="./Images/MAplot.png" width="350px" /> ] .pull-right[ <img src="./Images/Volcanoplot.png" width="350px" /> ] --- .center[ ### ¿Cuántos tipos de gráficos conoces para visualizar resultados del análisis DE? ] .pull-left[ <img src="./Images/MAplot.png" width="350px" /> **.green[MAplot]** ] .pull-right[ <img src="./Images/Volcanoplot.png" width="350px" /> **.green[Volcano plot]** ] --- # MAplot .scroll-output[ Gráfico que representa la distribución de los genes o transcritos en las comparaciones de interés **M** eje y de *minus*: .content-box-blue[$$logTx - logCT = logTx/CT$$ ] **A** eje x de *average* -> Promedio de las cuentas normalizadas para cada gen en todas las muestras Para generar el **MAplot** usemos el siguiente comando: .code90[ ```r ##Es importante que recuerden que la hipótesis nula que se probó fue ##"El lfc del gen n es igual a 0" por lo tanto los genes coloreados son... plotMA(res) ``` ] ] --- # MAplot <img src="Slides_files/figure-html/unnamed-chunk-119-1.png" width="450px" style="display: block; margin: auto;" /> --- # MAplot Para tener una mejor estimación del LFC de genes que: - Son poco abundantes - Tienen alta dispersión Usemos la función `lfcShrink` de la librería *.red[apeglm]* ```r ##En el argumento coef indicar el contraste entre los grupos experimentales res_lfc <- lfcShrink(dds, coef = "Treatment_Verafinib_vs_Control", type = "apeglm") ##Si desconocemos el nombre del contraste, usar: resultsNames(dds) ``` --- # MAplot .pull-left[ Sin `lfcshrink` <img src="Slides_files/figure-html/unnamed-chunk-121-1.png" width="450px" style="display: block; margin: auto;" /> ] .pull-right[ Con `lfcshrink` <img src="Slides_files/figure-html/unnamed-chunk-122-1.png" width="450px" style="display: block; margin: auto;" /> ] --- # MAplot .scroll-output[ También pueden generar un MAplot interactivo con la librería de *Glimma* ```r ##Para los objetos creados con DESeq2 es importante que incluyas un nivel llamado group group <- info$Treatment dds$group <- group glimmaMA(dds) ``` ] --- # Volcano plot De manera similar al MAplot con el volcano plot visualizamos los genes que muestran expresión diferencial en nuestra condición de interés - En el eje y se grafica el -log10 de padj - En el eje x se grafica el lfc o *log2foldchange* .code80[ ```r ##Para crear un volcano plot necesitas convertir los resultados de DESeq a un data frame DEG <- as.data.frame(res) ##En el script de funciones encontrarás la función de volcanoplotR para generar tu gráfico #los valores de los argumentos logfc y p.adj deben ser iguales al threshold utilizado para generar los resultados #en type debes indicar que los resultados provienen de DESeq volcanoplotR(DEG, logfc = 0, p.adj = 0.1, type = "DESeq") ``` ] --- # Volcano plot <img src="Slides_files/figure-html/unnamed-chunk-125-1.png" width="450px" style="display: block; margin: auto;" /> --- # Heatmap El *heatmap* nos permite visualizar la expresión de los genes diferencialmente expresados en terminos de las cuentas normalizadas Consideraciones: - Usar los valores de las cuentas normalizadas para una mejor comparación entre muestras - Escalar los valores de las cuentas (renglones) para visualizar las diferencias en la expresión Usaremos la librería de *.red[pheatmap]* --- # Heatmap .code90[ ```r ##Guardar la lista de transcritos que mostraron expresión diferencial significativa significant <- DEG %>% filter(log2FoldChange > 0 & padj < 0.1 | log2FoldChange < 0 & padj < 0.1) ##Generar la matriz de cuentas normalizadas norm_counts <- counts(dds, normalized = T) ##Cortar la matriz de cuentas normalizadas con los id de los transcritos diferencialmente expresados norm_counts <- norm_counts[rownames(significant), ] ##Generar una tabla de anotaciones preservando el tratamiento y el tipo de células annotation_col <- coldata[, c(2, 3)] ##Generar el heatmap empleando clustering jerarquico pheatmap(norm_counts, border_color = NA, scale = "row", clustering_distance_rows = "euclidean", clustering_distance_cols = "euclidean", clustering_method = "average", show_colnames = F, show_rownames = F, annotation_col = annotation_col) ``` ] --- # Heatmap <img src="Slides_files/figure-html/unnamed-chunk-127-1.png" width="450px" style="display: block; margin: auto;" />